w-转氨酶分子改造研究进展
2018-07-30高新星韦平和
高新星,韦平和
w-转氨酶分子改造研究进展
高新星,韦平和
泰州学院 医药与化学化工学院 江苏省手性医药化学品生物制造重点建设实验室,江苏 泰州 225300

金城. 2018酶工程专刊序言. 生物工程学报, 2018, 34(7): 1021−1023.Jin C. Preface for special issue on enzyme engineering (2018). Chin J Biotech, 2018, 34(7): 1021−1023.
w-转氨酶能催化羰基化合物发生不对称还原胺化反应,在制备手性胺类化合物方面具有较好的应用前景。由于底物结合区域特殊的空间结构,野生型w-转氨酶在合成大位阻手性胺方面的应用受到了限制。此外,在立体选择性和稳定性方面这一类酶也存在一些不足,目前满足工业应用需求的w-转氨酶仍较为有限。文中首先介绍了w-转氨酶的结构特征和催化机制,并探讨S型和R型酶在结构特征方面的主要差异。然后对w-转氨酶的分子改造研究进行了综述,重点阐述了基于结构特征和催化机制进行的分子改造研究,包括底物特异性改造、立体选择性改造和稳定性改造三方面。最后,对w-转氨酶分子改造研究进展进行总结和展望。
w-转氨酶,结构,分子改造,底物特异性,稳定性
手性胺是手性药物的重要组成部分,随着手性药物市场的不断扩大,手性胺的需求迅速增长。w-转氨酶能催化羰基化合物不对称还原胺化生成手性胺,反应过程中将氨基供体 (如D/L-丙氨酸或异丙胺) 的氨基转移至氨基受体 (羰基化合物),生成手性胺及副产物 (如丙酮酸或丙酮),其中氨基供体或氨基受体通常至少有一个不是a-氨基酸或a-酮酸[1-2]。利用w-转氨酶不对称催化合成手性胺能够避免现有手性胺合成技术存在的缺陷,如重金属污染、立体选择性差、产率低等,因而受到国内外学者的广泛关注和深入研究[3-6]。
w-转氨酶在合成手性胺方面具有较好的应用前景,但由于野生型酶在底物特异性、稳定性、催化效率等方面存在诸多不足,目前满足工业应用需求的w-转氨酶仍较为有限。基于非理性、理性及半理性设计策略的蛋白质工程技术能有效改善w-转氨酶的应用性能,为手性胺的高效制备提供了可能。随着w-转氨酶蛋白结构和催化机制相关研究的深入,利用理性或半理性设计策略对w-转氨酶进行分子改造的研究备受关注。文中主要对w-转氨酶的结构特征、催化机制及分子改造研究进行概述,以期为开发满足工业应用需求的w-转氨酶提供理论依据。
1 w-转氨酶的结构及催化机制
1.1 w-转氨酶的蛋白结构
w-转氨酶通常是由相同亚基组成的二聚体或四聚体,活性中心位于两个亚基的交界面,包括5′-磷酸吡哆醛 (Pyridoxal-5-phosphate,PLP) 结合区域、底物结合区域及关键催化残基 (赖氨酸)。根据空间位置和形状的不同,底物结合区域可分为两个结合口袋:大口袋和小口袋,分别用于结合底物结构中羰基或氨基两侧的基团,其中大口袋可以结合底物结构中的芳环、脂肪链以及羧基等体积较大或带电性基团,而小口袋由于空间位阻、带电性及疏水性等因素的影响只能容纳体积较小的基团 (如甲基)。
根据手性选择性的不同,w-转氨酶可分为S型和R型,其中S型w-转氨酶能以L-丙氨酸为氨基供体通常生成S型手性胺,而R型w-转氨酶能以D-丙氨酸为氨基供体通常生成R型手性胺。在PLP依赖型酶的分类中,S型和R型w-转氨酶分别属于折叠类型Ⅰ类和Ⅳ类[7-9],两者在结构方面既有相似之处又有明显差别。
来源于河流弧菌的w-转氨酶 (AT) 属于S型酶,有关该酶的研究报道较多,其晶体结构 (图1A) 已被解析[10-11]。该酶底物结合区域位于两个亚基的交界面,其中小口袋由两个亚基的8个位点组成,包括19Phe (A亚基)、150Tyr (A亚基)、165Tyr (A亚基)、85Phe (B亚基)、86Phe (B亚基)、320Gly (B亚基)、321Phe (B亚基)、322Thr (B亚基),而大口袋由A亚基上不同位点组成,包括57Trp、228Ala、258Val、259Ile、415Arg。活性中心关键催化残基285Lys位于PLP的面 (相对于C4′)。相比S型w-转氨酶而言,R型w-转氨酶的发现和研究起步则较晚。来源于土曲霉的w-转氨酶 (AT) 属于R型酶,其晶体结构 (图1B) 已被解析[12]。在两个亚基交界面的底物结合区域中,小口袋由A亚基上不同位点组成,包括62Val、274Thr、275Thr、276Ala,而大口袋由两个亚基的7个位点组成,包括60Tyr (A亚基)、115Phe (A亚基)、117Glu (A亚基)、182Leu (A亚基)、184Trp (A亚基)、55His (B亚基)、128Arg (B亚基)。活性中心关键催化残基180Lys位于PLP的面 (相对于C4′)。S型和R型w-转氨酶结构中关键催化残基 (Lys) 相对辅酶PLP或结合底物的空间位置成镜像关系,这是引起酶立体选择性差异的关键因素。
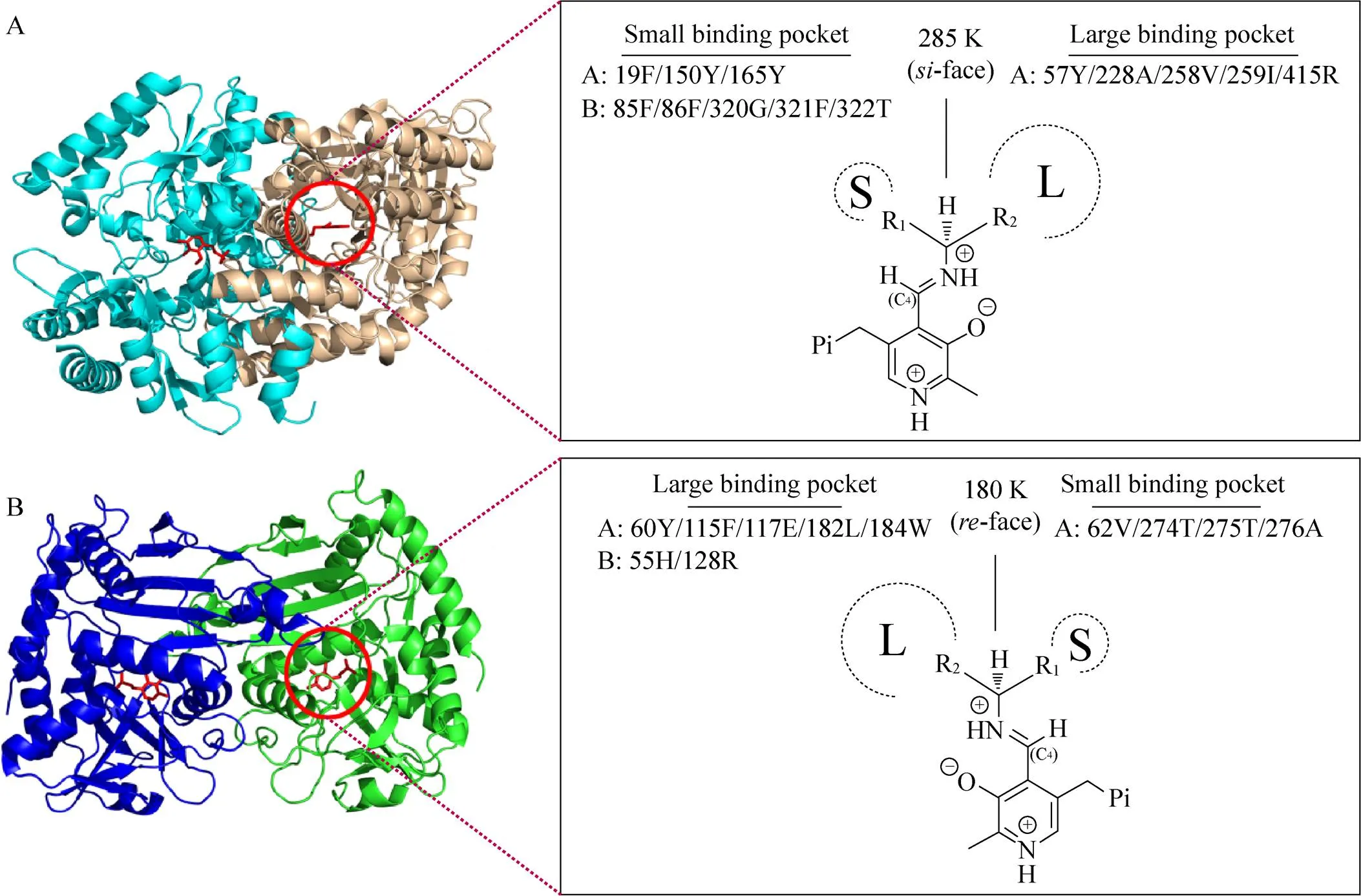
图1 w-转氨酶结构及活性中心示意图
1.2 w-转氨酶的催化机制
w-转氨酶属于PLP依赖型转移酶,其催化的转胺过程由两步可逆反应组成,遵循双底物乒乓反应机理[13-14],催化机制如图2所示。首先活性中心催化残基Lys的氨基与PLP的醛基通过形成内部醛亚胺 (Ⅰ) 的形式相连,然后由氨基供体的氨基取代Lys的氨基与PLP形成一个外部醛亚胺 (Ⅱ);在催化残基Lys作用下发生质子转移,先后形成醌型结构中间体 (Ⅲ) 和酮亚胺中间体 (Ⅳ);最后由酮亚胺中间体水解生成产物酮和5′-磷酸吡哆胺 (PMP)。第二步反应将PMP的氨基转移至潜手性酮上生成手性胺,同时完成辅酶循环再生。这一步反应中当催化残基Lys位于醌型结构中间体的面 (相对于PLP的C4′) 时 (S型转氨酶) 主要生成S型手性胺,而位于面时 (R型转氨酶) 主要生成R型手性胺。
2 w-转氨酶的分子改造
野生型w-转氨酶在不对称催化合成手性胺方面存在诸多不足,通过分子改造研究能有效提升w-转氨酶的催化特性,主要包括底物特异性、立体选择性及稳定性三方面。
2.1 w-转氨酶的底物特异性改造
2.1.1w-转氨酶的底物偏好性改造
根据催化底物类型的不同,转氨酶可分为a-转氨酶和w-转氨酶。与a-转氨酶不同,w-转氨酶的底物范围不仅限于a-氨基酸或a-酮酸,还包括酮类和醛类化合物[15-16]。由于底物结合区域空间结构的限制,尤其是小口袋只能容纳体积较小的基团,野生型w-转氨酶最适反应底物通常为a-酮酸或甲基酮类化合物,但对于不同结构的a-酮酸或甲基酮类化合物,其催化活性差别也较大。通过探究影响w-转氨酶底物偏好性的主要因素,并在此基础上开展的底物偏好性改造研究取得一定进展。

图2 w-转氨酶的催化机制
w-转氨酶对脂肪胺类底物的催化活性通常比芳香胺类底物要低得多,Cho等[17]以AT为研究对象,通过同源建模和构效关系分析发现底物结合区域大口袋中第57位和147位的色氨酸影响酶与脂肪胺类底物的结合,将其突变为侧链基团较小的甘氨酸后,两个突变体对不同结构的脂肪胺和芳香胺均展现出更好的催化活性,其中对苯基- 4-丁胺的催化活性分别提高至野生型酶的40.6倍和14.3倍,对6-甲基-2-庚胺的催化活性分别提高至野生型酶的14.6倍和1.4倍。来源于新月柄杆菌的S型w-转氨酶与AT具有相似的底物特异性,Hwang等[18]将该酶底物结合区域第227位缬氨酸和285位天冬酰胺分别突变为侧链较小的甘氨酸和丙氨酸后,酶底物特异性发生明显改变。两种突变体对3-氨基-3-苯基丙酸的催化活性分别提高了1.4倍和1.9倍,而对3-氨基丁酸的催化活性降低至野生型酶的56%和28%。嗜盐单胞菌来源的S型w-转氨酶能够催化不同结构的芳环类底物,Contente等[19]结合计算机辅助设计和实验检测分析手段探究底物结合区域立体电子效应对该酶催化芳环类底物的影响,将底物结合区域第56位色氨酸突变为甘氨酸后,降低了立体电子效应和空间位阻效应对酶结合芳环类底物的影响,突变体W56G对邻硝基苯乙酮的催化活性较野生型酶提高2倍以上,同时对苯乙酮、间硝基苯乙酮、对硝基苯乙酮的催化活性较野生型酶未降低。空间位阻效应是影响w-转氨酶底物偏好性的重要因素,将影响酶与底物相互作用的氨基酸残基突变为侧链基团更小的氨基酸残基,能够有效拓宽底物结合区域的空间,进而改变酶的底物偏好性。
尽管w-转氨酶的底物谱相对较广,然而对于酮类和醛类化合物的催化活性通常要远远低于其天然底物丙酮酸,这也限制了w-转氨酶在合成手性胺方面的应用。为提高w-转氨酶对不同结构酮类化合物的催化活性,Han等[20]对人苍白杆菌来源的S型w-转氨酶 (AT) 的底物偏好性进行改造,利用丙氨酸扫描技术对酶活性中心附近 (<3 Å) 的8个氨基酸残基进行突变,发现突变体W58A的催化活性较野生型酶明显提高。通过对该位点进行饱和突变和筛选,获得的单点突变体W58L对不同结构酮类底物的催化活性分别提高至野生型酶的41−760倍,而对丙酮酸的催化效率降至野生型酶的0.41倍。Genz等[21]基于同源序列比对和蛋白结构分析结果对AT的底物特异性改造进行半理性设计,在底物结合区域的大口袋中,Leu56、Trp57、Arg415、Leu417四个位点对酶结合a-酮酸或醛的选择性有重要影响,通过组合突变和筛选获得不同突变体的底物偏好性由丙酮酸转变为戊醛,在分别以 (S)-1-苯基乙胺、(S)-1-苯基丙胺或 (S)-1-苯基丁胺为氨基供体催化戊醛转胺的反应中,突变体M3 (W57F、R415L、417V) 的酶活提高至野生型酶的2.8、3.0、5.1倍。Han等[22]发现AT对于a-酮酸和醛类底物的催化活性要远远高于大部分潜手性酮类底物,基于蛋白结构特征和酶催化机制分析发现潜手性酮与酶分子的结合取向不利于反应过程中的亲核攻击,从而导致催化效率较低。在此基础上,结合计算机辅助分析和虚拟筛选技术开发出一种活性中心改造策略,利用该策略获得双点突变体 (L57A、W58A) 对苯乙酮、苯丙酮及苯丁酮的催化活性较野生型酶分别提高188倍、2 500倍和130 000倍。本研究团队发现AT对4-羟基丁酮的催化活性远远低于天然底物丙酮酸。利用同源建模和底物对接技术确定该酶底物结合区域内影响酶与底物相互作用的关键位点,通过单点饱和突变和多点组合突变,获得不同的突变体对4-羟基丁酮的催化活性较野生型酶至少提高2倍以上,其中突变体H55A/S215P底物结合区域较野生型酶发生明显变化 (图3),在不对称催化合成 (R)-3-氨基丁醇的反应中,底物浓度高达50 g/L,具有较好的工业应用前景[23]。酮类和醛类化合物是w-转氨酶不对称催化合成手性胺的重要原料,提高w-转氨酶对这一类化合物的催化活性能够更好地推动w-转氨酶的工业化应用。
除了上述研究之外,通过分子改造使得w-转氨酶具备a-转氨酶催化特性的研究也引起了人们的兴趣。Deszcz等[24]结合蛋白结构比对、系统进化分析、酶与底物相互作用分析等方法对来源于紫色色杆菌的S型w-转氨酶(AT) 的底物特异性改造进行半理性设计,利用定点饱和突变技术对底物结合区域的14个位点进行突变,通过筛选发现第60位、第88位和第153位的部分单点突变体展现出更高的丝氨酸/丙酮酸催化活性,较野生型酶提高2−4倍。具备催化不同结构类型底物的能力,即“底物杂泛性”,是酶的另一重要特性[25]。通过分子改造研究使得w-转氨酶具备“底物杂泛性”,对w-转氨酶不对称催化合成手性胺的理论研究和应用研究均有重要意义。
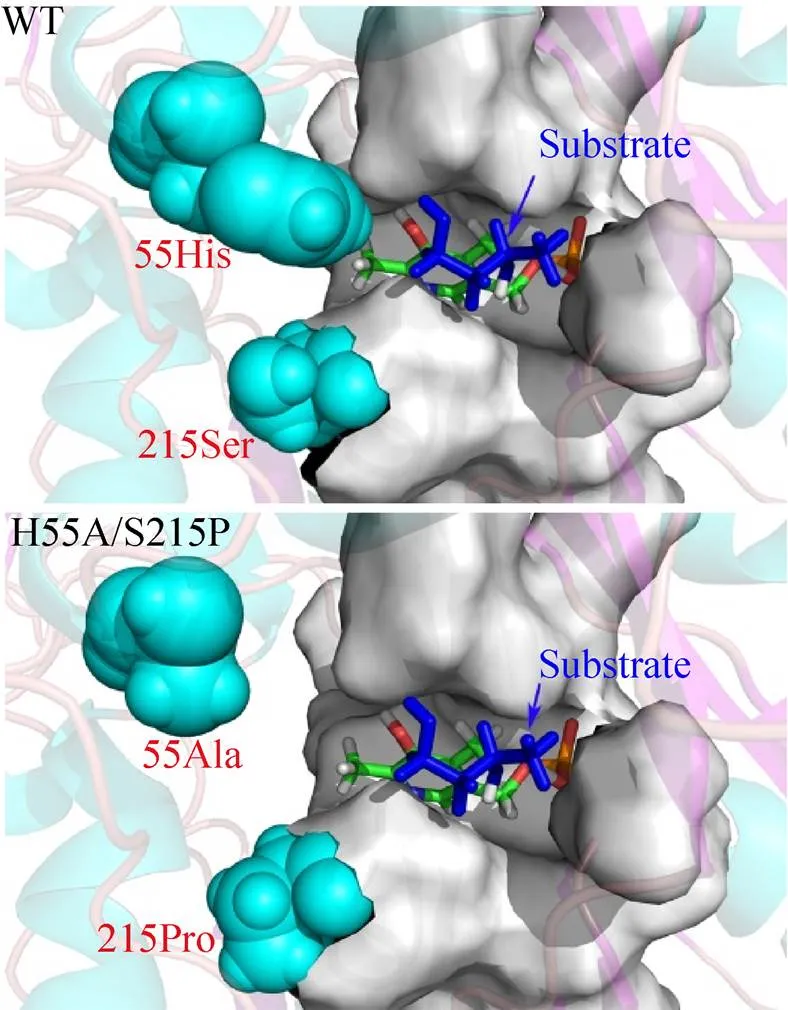
图3 AtAT及突变体H55A/S215P底物结合区域结构
2.1.2w-转氨酶对大位阻底物催化性能的改造
通过底物偏好性改造能有效改善w-转氨酶对a-酮酸、甲基酮类和醛类化合物的催化能力,然而作为药物中间体的手性胺通常在手性基团两侧含有较大的基团,对于这一类化合物,w-转氨酶通常无法合成或者效率极低,这也成为制约w-转氨酶不对称催化合成手性胺技术发展的主要瓶颈。影响w-转氨酶催化大位阻底物的关键因素是底物结合区域的空间结构,利用蛋白质工程技术手段对底物结合区域进行合理改造,能有效提高w-转氨酶对大位阻底物的催化性能,进而拓宽w-转氨酶在手性胺合成方面的应用空间。
在不同来源的w-转氨酶中,有关AT催化大位阻底物的分子改造研究最多。起初由于缺乏蛋白结构和催化机制等信息,人们主要采用非理性设计方法对AT进行改造。Hwang等[26]利用易错PCR技术对目的基因进行随机突变,以3-氨基-3-苯基丙酸为唯一氮源进行初筛培养,然后通过反应过程中生成的a-氨基酸与铜离子发生的显色反应进行高通量筛选,获得不同突变体对3-氨基-3-苯基丙酸的转化率提高至野生型酶的1.3−3.1倍。Yun等[15]利用易错PCR技术构建突变体库,以2-氨基庚烷作为唯一氮源进行富集培养,同时添加丁酮作为转氨酶的抑制产物,通过逐步提高丁酮浓度进行筛选,获得的突变体w-Tamla (P233L、V297A) 对2-氨基庚烷、2-氨基- 6-甲基庚烷及2-氨基辛烷的催化活性分别提高至野生型酶的1.7−2倍,同时对产物丁酮和2-庚酮的抑制常数分别提高至野生型酶的6倍和4.5倍。尽管非理性改造无需事先了解目的蛋白的空间结构和催化机制,但研究过程中需结合有效的高通量筛选方法对数量庞大的突变体库进行筛选,耗时耗力。
随着AT蛋白结构和催化机制相关研究的深入,基于理性或半理性设计策略的分子改造研究日益增多。在催化 (R)-5-甲基-3-氧代辛酸乙酯合成 (3S,5R)-3-氨基-5-甲基辛酸乙酯的分子改造研究中,Midelfort等[11]基于同源序列比对、结构分析及ProteinGPSTM统计分析等方法将突变位点限制在AT活性中心附近,经过4轮突变共构建约450个突变体,经筛选获得最佳突变体r414 (F19W、W57F、F85A、R88K、V153A、K163F、I259V、R415F) 的催化活性较野生型酶提高60倍。为进一步提高分子改造效率,Sirin等[27]结合分子动力学模拟、分子对接及分子力学评分等方法开发了一种可以预测不同突变对酶催化活性影响的计算机程序,利用该程序对AT进行理性设计,有效提高了实验效率,从构建的89个不同突变体中获得两个最佳突变体,催化活性较野生型酶分别提高20和60倍,其中包括上述研究中获得的突变体r414。此外,AT还被用于催化其他不同结构大位阻底物的研究,Dourado等[28]利用蛋白结构分析、分子对接、分子动力学模拟、量子力学计算、计算机辅助蛋白结构稳定性研究、共进化网络分析及体外筛选等技术手段对AT进行理性分子改造,共构建113个不同突变体,获得最佳突变体有7个位点发生突变 (W57F、R88H、V153S、K163F、I259M、R415A、V422A),在不对称催化合成 (1S)-1-(2-联苯) 乙胺的反应中反应速率较野生型酶提高1 716倍。Nobili等[10]发现AT对于含有大位阻基团的a-羟基酮类和芳基-烷基酮类底物的催化活性较低,将底物结合区域小口袋内的部分位点进行单点饱和突变和组合突变,通过筛选获得突变体F85L/V153A对 (S)-苯丁胺的催化活性是野生型酶的26倍,突变体Y150M/V153A对 (R)-苯甘氨醇的催化活性是野生型酶的53倍。Genz等[29]将AT底物结合区域中的7个不同位点进行组合突变,在拓宽结合口袋空间的同时保持其疏水环境,通过筛选获得不同突变体能够催化含有支链烷烃结构的手性胺,其中突变体H3_RAV (L56V、W57C、F85V、V153A) 在不对称催化合成 (R)-2,2-二甲基-1-苯丙胺反应中具有较好的催化活性和立体选择性,而野生型酶无明显催化活性。
除AT之外,有关其他来源的w-转氨酶的分子改造研究亦较多。来源于脱氮副球菌的S型w-转氨酶底物结合区域的小口袋能够容纳体积较大的基团 (如丁基或丙基),Park等[30]对该酶底物结合区域小口袋中6个不同位点进行丙氨酸扫描突变分析,发现突变体V153A对不同结构大位阻底物均展现出一定的催化活性,在以2-氧代辛酸和 (S)-苯乙胺为底物的反应中,突变体V153A的催化活性较野生型酶提高3倍。Martin等[31]以来源于柠檬节杆菌的S型w-转氨酶 (AT) 为研究对象,利用易错PCR构建突变体库,以 (S)-2-四氢萘胺类化合物和丙酮酸为筛选反应底物,产物2-四氢萘酮类化合物接触空气易发生颜色变化,根据反应溶液颜色深浅可判断酶活高低,从而实现高通量筛选。经过5轮突变和筛选获得的突变体有17个位点发生突变,催化活性较野生型酶提高268倍,能满足 (S)-2-四氢萘胺类化合物的工业生产需求。2010年美国总统绿色化学挑战奖的绿色反应条件奖授予了Merck和Codexis公司,以表彰它们在Ⅱ型糖尿病治疗药物-西他列汀 (Sitagliptin) 的绿色生产中所作的贡献,这也是利用w-转氨酶不对称催化合成手性胺技术发展的重要里程碑[32-33]。西他列汀潜手性酮在羰基两侧均含有大位阻基团,野生型酶对该化合物几乎无催化活性,Savile等[32]以节杆菌sp.来源的R型w-转氨酶ATA117为研究对象,首先利用定点饱和突变技术对底物结合区域大口袋进行改造,获得突变体1 (S223P) 对西他列汀潜手性酮结构类似物 (甲基酮) 展现一定的催化活性。然后结合定点饱和突变、组合突变及随机突变技术对底物结合区域小口袋及附近位点进行改造,获得突变体2 (S223P/Y26H/V65A/V69G/F122I/A284G)首次对西他列汀潜手性酮展现出催化活性。在此基础上将底物结合区域其他不同位点的正向突变进行组合,获得突变体3 (突变体2+H62T/G136Y/ E137I/V199I/A209L/T282S) 对西他列汀潜手性酮的催化活性较突变体2提高75倍,经过11轮突变最终获得突变体Rd11TA,其底物结合区域能有效结合西他列汀潜手性酮 (图4),催化活性较野生型酶提高了28 000倍以上。
不同来源的w-转氨酶在蛋白结构方面存在明显差异,针对单个酶的分子改造研究结果往往难以复制到其他来源的酶中。因此,系统研究w-转氨酶在催化含有大取代基底物方面的结构与功能关系,并确定影响酶与底物结合的关键因素,有助于推进w-转氨酶对大取代基底物的催化性能改造。Pavlidis等[7]基于酶与底物相互作用关系对鲁杰氏菌sp. TM1040来源的S型w-转氨酶进行理性改造,获得的突变体能够不对称催化合成一类大位阻手性胺,其中对1,3-二苯基- 1-丙胺的催化活性较野生型酶提高了8 900倍。在此基础上鉴定出底物结合区域中影响酶催化合成含有大位阻手性胺的关键基序 (Y59W、Y87F、Y152F、T213A、P423H),并验证该基序在不同来源的S型w-转氨酶中具有较好的适用性。基于上述研究,Weiß等[34]通过分子改造研究首次将w-转氨酶用于不对称催化合成双环胺类化合物,他们以上述研究中的突变体3FCR_QM作为起始框架,结合定点突变、饱和突变及易错PCR等方法构建突变体库,经筛选获得5个正向突变体,在不对称催化合成外向-3-氨基-8-氮杂双环[3.2.1]辛-8-基-苯基-甲酮的反应中的转化率为起始框架3FCR_QM的2.2−4.2倍。在另一项研究[35]中,该团队基于相同的策略对前期研究中构建的突变体3FCR_Y59W/T231A进行分子改造,获得突变体3FCR_WLAMA (Y59W、Y87L、T231A、L382M、G429A) 能够不对称催化2,2-二甲基苯丙酮合成 (R)-2,2-二甲基-1-苯丙胺,而原始突变体3FCR_Y59W/T231A无明显催化活性。
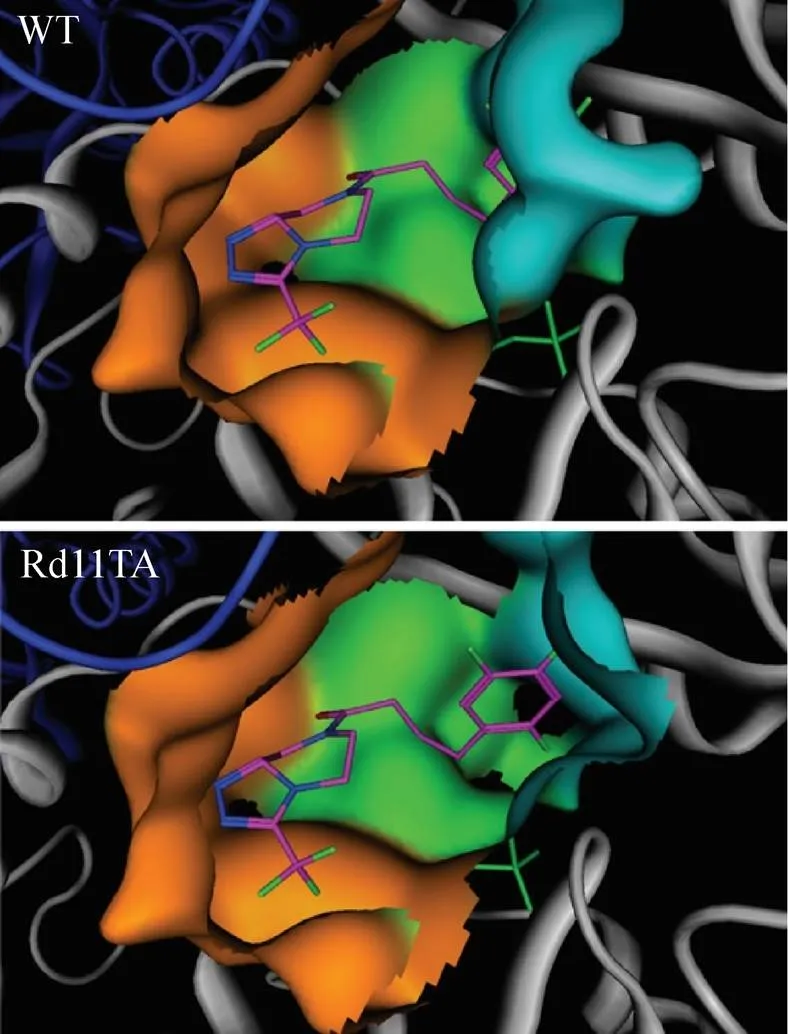
图4 节杆菌来源的w-转氨酶及突变体Rd11TA底物结合区域结构[32]
2.2 w-转氨酶的立体选择性改造
在w-转氨酶不对称催化过程中,生成何种构型的手性产物取决于底物在酶活性中心的结合取向。当底物的面 (相对于底物中的潜手性羰基碳原子) 朝向关键催化残基Lys时生成S型产物,反之则生成R型产物 (图5)。w-转氨酶通常具有较好的立体选择性,但部分结构特殊的底物在底物结合区域存在不同的结合取向,能生成不同构型的手性产物,从而影响产物的手性纯度。
通过对w-转氨酶底物结合区域空间结构的改造能够改变底物的结合取向,使其倾向于生成单一构型的手性产物,从而有效提高或转变酶的立体选择性。Svedendahl等[36]以突变体CNB05-01为研究对象,基于同源建模及蛋白结构分析推测活性中心的一个loop结构可能对酶的立体选择性存在影响,对该区域3个位点 (E326、V328、Y331) 进行定点突变研究,结果显示突变体CNB05-01/Y331C对4-氟苯基丙酮的立体选择性 (值) 由98% (S) 提高至99.5% (S),而突变体CNB05-01/V328A对4-氟苯基丙酮的立体选择性发生翻转,R型产物ee值达到58%。Skalden等[37]通过分析AT与反应中间体 (内部醛亚胺中间体) 的相互作用确定底物结合区域第56位亮氨酸影响酶催化的立体选择性,根据同源序列比对结果将突变目标氨基酸锁定为异亮氨酸、缬氨酸、丙氨酸、色氨酸、酪氨酸及苯丙氨酸,以 (S) - 3-甲基环己酮为底物对6种单点突变体检测,结果显示突变体L56V较野生型酶展现出更好的R型选择性,非对映体过剩值由12%提高至66%,而突变体L56I立体选择性则转变为S型,非对映体过剩值为71%。
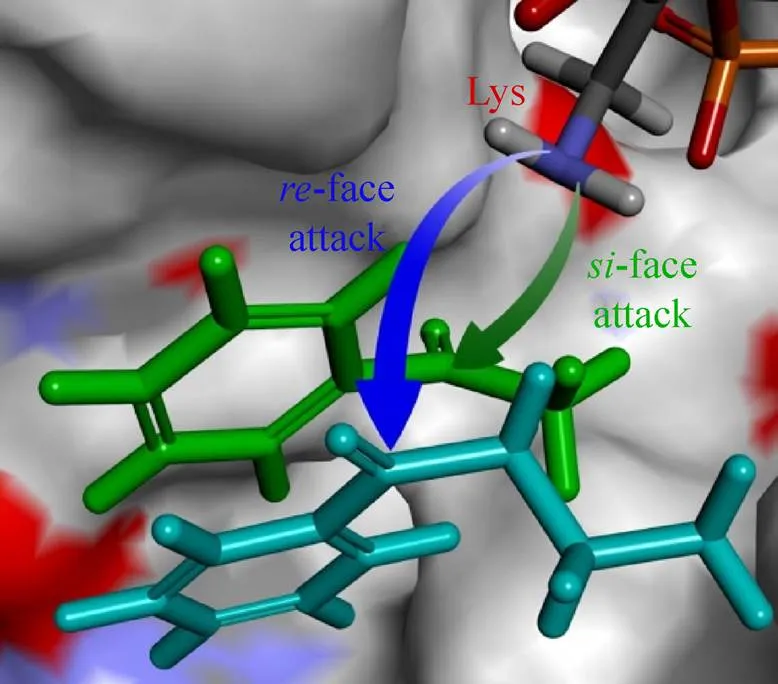
图5 w-转氨酶中底物结合取向示意图[22]
此外,Humble等[38]尝试不同策略对AT的立体选择性进行理性改造。首先,根据同源结构比对结果将底物结合区域第60位色氨酸突变为侧链基团较小的半胱氨酸,突变体W60C对甲基苄胺、1-四氢萘胺及2-四氢萘胺的对映体特异性 (E值) 较野生型酶分别提高1.3倍、7.9倍和14倍。然后,通过底物对接分析发现底物苯乙酮与酶的结合取向更倾向于生成S构型的产物,将结合底物两侧的位点F88和A231分别突变为丙氨酸和苯丙氨酸后,底物与酶的结合取向发生翻转,使其更倾向于生成R构型产物,结果显示突变体F88A/A231F对1-四氢萘胺的立体选择性 (E值) 分别由6.7 (S) 转变为1.9 (R),对2-四氢萘胺的立体选择性 (E值) 分别由3.9 (S) 转变为63 (R)。最后,基于蛋白结构和催化机制分析结果,将关键催化残基Lys与底物相对位置由面改为面,以期转变酶的立体选择性,然而由于突变体酶活丧失或下降严重并未取得理想结果。
从理论上讲,改变w-转氨酶活性中心关键催化残基与底物的相对位置 (面与面转换) 可以转变酶的立体选择性,但是这需要大范围改造酶活性中心的空间结构,单纯改变关键催化残基的位置极易导致酶活性下降或丧失。相比这种策略,通过改造酶底物结合区域来改变底物结合取向更能有效提高或转变酶的立体选择性。
2.3 w-转氨酶的稳定性改造
w-转氨酶不对称催化合成手性胺的过程中,通常需要承受高温 (提高酶催化速率及底物溶解性)、高盐 (高浓度氨基供体)、高浓度有机溶剂 (提高底物溶解性) 等反应条件。因此,提升w-转氨酶的稳定性具有重要意义。
酶分子的稳定性与蛋白结构的刚性有着密切联系,通常蛋白结构的刚性越强其维持活性构象的能力也越强,因此,合理增强酶分子柔性区域的刚性有助于提高酶分子的稳定性。Huang等[39]依据蛋白结构温度因子 (B因子) 和折叠自由能参数开发了一种提高酶温度稳定性的策略,利用该策略对AT进行半理性改造,通过增强结构中柔性区域 (Gly129-Asp134) 的刚性使得酶分子温度稳定性明显提高,获得的最佳突变体T130M/E133F由于分子内部疏水作用 (M130与I135、P132与F133) 和氢键作用 (M130与I135) 的增强 (图6),其稳定性明显提高,半衰期由6.9 min提高至22.7 min,解链温度由38.5 ℃提高至43.5 ℃。
影响酶分子稳定性的因素较多,包括蛋白结构柔性、酶分子内部疏水作用、亚基之间结合程度、蛋白表面带电性等,针对单一因素改善酶稳定性的效果往往并不理想。因此,结合多种影响因素的分子改造研究对提升酶稳定性有着重要作用。在w-转氨酶不对称催化合成西他列汀手性胺中间体的研究[32]中,通过对w-转氨酶ATA117的11轮突变和筛选最终获得的突变体 (Rd11TA) 包含27个突变点,其中活性中心附近的突变S223P能够稳定底物结合区域loop结构,位于蛋白结构表面的突变G215C有助于提高酶的稳定性,位于亚基交界面的突变L61Y、V65A、M94I、Y150S、V152C能够加强二聚体的相互作用。最终突变体Rd11TA在温度稳定性、有机溶剂耐受性以及高盐耐受性方面的能力大幅提升,在严苛的反应条件 (50% DMSO、1 mol/L异丙胺、250 g/L底物、反应温度45 ℃) 下仍能保持较好的催化活性,能够满足西他列汀手性胺中间体的生产制备需求。在近期一项关于荧光假单胞菌来源的S型w-转氨酶的稳定性改造研究中,Börner等[40]基于底物诱导酶失活的机理将突变区域限制在辅酶结合区域,对该区域32个位点进行定点饱和突变和组合突变,通过筛选获得的突变体ATA-v2 (S51G、N161I、Y164L) 由于分子内部氢键作用和疏水作用的增强而在有机溶剂耐受性、异丙胺耐受性、温度稳定性及催化效率等方面展现出更好的性能。
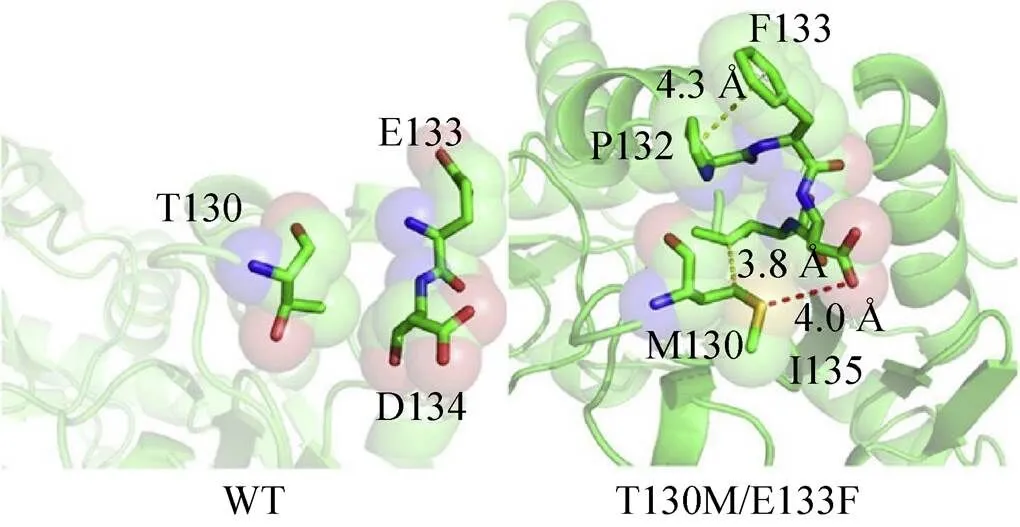
图6 AtAT及突变体T130M/E133F分子内部相互作用模型分析[39]
3 总结与展望
随着w-转氨酶相关研究的深入,人们对其催化特性、结构特征及催化机制有着较清晰的认识,在此基础上进行的w-转氨酶分子改造研究也取得了快速发展。一方面,有关w-转氨酶分子改造的设计理念发生转变,由建立在大量筛选工作基础之上的非理性设计向基于蛋白结构功能关系和计算机辅助设计的理性或半理性设计转变,使得酶分子改造的目的性更强、有效性更高。另一方面,在关于w-转氨酶底物特异性的分子改造研究中,目标化合物由结构简单、接近天然底物 (甲基酮或a-酮酸) 结构的潜手性酮向结构复杂、附加值更高的药物中间体潜手性酮转变,从而进一步拓宽w-转氨酶在手性药物制造行业的应用空间。
尽管有关w-转氨酶分子改造的研究较多,然而大部分研究尚处于实验室阶段,能够满足工业化生产需求的案例并不多见。因此,拓宽w-转氨酶的应用空间并推动其工业化应用将成为w-转氨酶分子改造研究的努力方向。在文中概述的26篇分子改造研究中,针对R型w-转氨酶的研究仅有3篇,加强R型w-转氨酶的分子改造研究,尤其是改善对大位阻底物催化性能的研究,能够拓宽w-转氨酶在R型手性胺制备方面的应用空间。此外,酶分子改造过程中不可避免地涉及大量挑选和筛选工作,充分利用自动化技术,如高通量菌落挑选仪、自动移液工作站等设备,将大大提高工作效率,缩短酶分子改造周期。随着基因组学、结构生物学、生物信息学等技术的迅速发展,基于蛋白序列-结构-功能关系和计算机辅助设计驱动的理性或半理性改造技术将会更加成熟,充分结合非理性、理性及半理性设计的优势有助于提升w-转氨酶分子改造研究成效,加快推进w-转氨酶在手性药物工业化生产中的应用。
[1] Koszelewski D, Tauber K, Faber K, et al.w-Transaminases for the synthesis of non-racemica-chiral primary amines. Trends Biotechnol, 2010, 28(6): 324–332.
[2] Steffen-Munsberg F, Vickers C, Kohls H, et al. Bioinformatic analysis of a PLP-dependent enzyme superfamily suitable for biocatalytic applications. Biotechnol Adv, 2015, 33(5): 566–604.
[3] Hollmann F, Arends IWCE, Holtmann D. Enzymatic reductions for the chemist. Green Chem, 2011, 13(9): 2285–2314.
[4] Fuchs M, Farnberger JE, Kroutil W. The industrial age of biocatalytic transamination. Eur J Org Chem, 2015, 2015(32): 6965–6982.
[5] Mathew S, Yun H.w-Transaminases for the production of optically pure amines and unnatural amino acids. ACS Catal, 2012, 2(6): 993–1001.
[6] Guo F, Berglund P. Transaminase biocatalysis: optimization and application. Green Chem, 2017, 19(2): 333–360.
[7] Pavlidis IV, Weiß MS, Genz M, et al. Identification of (S)-selective transaminases for the asymmetric synthesis of bulky chiral amines. Nat Chem, 2016, 8(11): 1076–1082.
[8] Höhne M, Schätzle S, Jochens H, et al. Rational assignment of key motifs for function guidesenzyme identification. Nat Chem Biol, 2010, 6(11): 807–813.
[9] Percudani R, Peracchi A. The B6 database: a tool for the description and classification of vitamin B6-dependent enzymatic activities and of the corresponding protein families. BMC Bioinform, 2009, 10: 273.
[10] Nobili A, Steffen-Munsberg F, Kohls H, et al. Engineering the active site of the amine transaminase fromfor the asymmetric synthesis of aryl-alkyl amines and amino alcohols. ChemCatChem, 2015, 7(5): 757–760.
[11] Midelfort KS, Kumar R, Han S, et al. Redesigning and characterizing the substrate specificity and activity ofaminotransferase for the synthesis of imagabalin. Protein Eng Des Sel, 2013, 26(1): 25–33.
[12] Łyskowski A, Gruber C, Steinkellner G, et al. Crystal structure of an (R)-selective ω-transaminase from. PLoS ONE, 2014, 9(1): e87350.
[13] Cassimjee KE, Humble MS, Miceli V, et al. Active site quantification of an ω-transaminase by performing a half transamination reaction. ACS Catal, 2011, 1(9): 1051–1055.
[14] Slabu I, Galman JL, Lloyd RC, et al. Discovery, engineering and synthetic application of transaminase biocatalysts. ACS Catal, 2017, 7(12): 8263–8284.
[15] Yun H, Hwang BY, Lee JH, et al. Use of enrichment culture for directed evolution of theJS17w-transaminase, which is resistant to product inhibition by aliphatic ketones. Appl Environ Microbiol, 2005, 71(8): 4220–4224.
[16] Malik MS, Park ES, Shin JS. Features and technical applications of ω-transaminases. Appl Microbiol Biotechnol, 2012, 94(5): 1163–1171.
[17] Cho BK, Park HY, Seo JH, et al. Redesigning the substrate specificity ofw-aminotransferase for the kinetic resolution of aliphatic chiral amines. Biotechnol Bioeng, 2008, 99(2): 275–284.
[18] Hwang BY, Ko SH, Park HY, et al. Identification of ω-aminotransferase fromand site-directed mutagenesis to broaden substrate specificity. J Microbiol Biotechnol, 2008, 18(1): 48–54.
[19] Contente ML, Planchestainer M, Molinari F, et al. Stereoelectronic effects in the reaction of aromatic substrates catalysed bytransaminase and its mutants. Org Biomol Chem, 2016, 14(39): 9306–9311.
[20] Han SW, Park ES, Dong JY, et al. Mechanism-guided engineering of ω-transaminase to accelerate reductive amination of ketones. Adv Synth Catal, 2015, 357(8): 1732–1740.
[21] Genz M, Vickers C, van den Bergh T, et al. Alteration of the donor/acceptor spectrum of the (S)-amine transaminase from. Int J Mol Sci, 2015, 16(11): 26953–26963.
[22] Han SW, Kim J, Cho HS, et al. Active site engineering of ω-transaminase guided by docking orientation analysis and virtual activity screening. ACS Catal, 2017, 7(6): 3752–3762.
[23] Gao XX, Lu LY, Zhu W. An improved transaminase and its application in the preparation of (R)-3-aminobutan-1-ol: CN, 2016111815359. 2016-12-20 (in Chinese). 高新星, 陆丽英, 竺伟. 一种改进的转氨酶及其在(R)-3-氨基丁醇制备上的应用: 中国, 2016111815359.2016-12-20.
[24] Deszcz D, Affaticati P, Ladkau N, et al. Single active-site mutants are sufficient to enhance serine: pyruvatea-transaminase activity in an ω-transaminase. FEBS J, 2015, 282(13): 2512–2526.
[25] Humble MS, Berglund P. Biocatalytic promiscuity. Eur J Org Chem, 2011, 2011(19): 3391–3401.
[26] Hwang BY, Kim BG. High-throughput screening method for the identification of active and enantioselectivew-transaminases. Enzyme Microb Technol, 2004, 34(5): 429–436.
[27] Sirin S, Kumar R, Martinez C, et al. A computational approach to enzyme design: predicting ω-aminotransferase catalytic activity using docking and MM-GBSA scoring. J Chem Inf Model, 2014, 54(8): 2334–2346.
[28] Dourado DFAR, Pohle S, Carvalho ATP, et al. Rational design of a (S)-selective-transaminase for asymmetric synthesis of (1S)-1-(1,1′-biphenyl-2-yl) ethanamine. ACS Catal, 2016, 6(11): 7749–7759.
[29] Genz M, Melse O, Schmidt S, et al. Engineering the amine transaminase fromtowards branched-chain substrates. ChemCatChem, 2016, 8(20): 3199–3202.
[30] Park ES, Park SR, Han SW, et al. Structural determinants for the non-canonical substrate specificity of the ω-transaminase from. Adv Synth Catal, 2014, 356(1): 212–220.
[31] Martin AR, Disanto R, Plotnikov I, et al. Improved activity and thermostability of (S)-aminotransferase by error-prone polymerase chain reaction for the production of a chiral amine. Biochem Eng J, 2007, 37(3): 246–255.
[32] Savile CK, Janey JM, Mundorff EC, et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science, 2010, 329(5989): 305–309.
[33] Desai AA. Sitagliptin manufacture: a compelling tale of green chemistry, process intensification, and industrial asymmetric catalysis. Angew Chem Int Edit, 2011, 50(9): 1974–1976.
[34] Weiß MS, Pavlidis IV, Spurr P, et al. Protein-engineering of an amine transaminase for the stereoselective synthesis of a pharmaceutically relevant bicyclic amine. Org Biomol Chem, 2016, 14(43): 10249–10254.
[35] Weiß MS, Pavlidis IV, Spurr P, et al. Amine transaminase engineering for spatially bulky substrate acceptance. ChemBioChem, 2017, 18(11): 1022–1026.
[36] Svedendahl M, Branneby C, Lindberg L, et al. Reversed enantiopreference of an ω-transaminase by a single-point mutation. ChemCatChem, 2010, 2(8): 967–980.
[37] Skalden L, Peters C, Dickerhoff J, et al. Two subtle amino acid changes in a transaminase substantially enhance or invert enantiopreference in cascade syntheses. ChemBioChem, 2015, 16(7): 1041–1045.
[38] Humble MS, Cassimjee KE, Abedi V, et al. Key amino acid residues for reversed or improved enantiospecificity of an ω-transaminase. ChemCatChem, 2012, 4(8): 1167–1172.
[39] Huang J, Xie DF, Feng Y. Engineering thermostable (R)-selective amine transaminase fromthroughdesign employing B-factor and folding free energy calculations. Biochem Biophys Res Commun, 2017, 483(1): 397–402.
[40] Börner T, Rämisch S, Bartsch S, et al. Three in one go: thermo-, solvent and catalytic stability by engineering the cofactor-binding element of amine transaminases. ChemBioChem, 2017, 18(15): 1482–1486.
(本文责编 陈宏宇)
Advances in molecular modification ofw-transaminase
Xinxing Gao, and Pinghe Wei
Jiangsu Key Laboratory of Chiral Pharmaceuticals Biosynthesis, College of Pharmacy and Chemistry and Chemical Engineering, Taizhou University, Taizhou 225300, Jiangsu, China
w-Transaminase catalyzes the asymmetric reductive amination of carbonyl compounds, and has great application prospect in the preparation of chiral amines. The application in synthesis of bulky chiral amines is limited by the special structure of substrate binding region in the wild-type enzyme. Moreover, there are also some drawbacks in the stereoselectivity and stability ofw-transaminase. So far,w-transaminase satisfying the industrial requirements is still rare. In this review, we first introduce the structure and catalytic mechanism ofw-transaminase, and then discuss the structural differences between S-selective and R-selective enzymes. Molecular modification ofw-transaminase was introduced in detail, by focusing on the structure and mechanism-based molecular modification, including substrate specificity, stereoselectivity, and stability.
w-transaminase, structure, molecular modification, substrate specificity, stability
November 20, 2017;
January 16, 2018
Scientific Research Starting Foundation of Taizhou University (No. TZXY2017QDJJ008), Open Foundation of Jiangsu Key Laboratory of Chiral Pharmaceuticals Biosynthesis, Taizhou University (No. SX1701).
10.13345/j.cjb.170455
Pinghe Wei. Tel/Fax: +86-523-80769295; E-mail: phwei2011@126.com
泰州学院科研启动基金项目 (No. TZXY2017QDJJ008),江苏省手性医药化学品生物制造重点建设实验室 (泰州学院) 开放课题基金 (No. SX1701) 资助。
