利用CRISPR/Cas9基因组编辑技术定向降低水稻落粒性
2018-07-25盛夏冰谭炎宁孙志忠余东汪雪峰袁贵龙袁定阳段美娟
盛夏冰,谭炎宁,孙志忠,余东,汪雪峰,袁贵龙,袁定阳,段美娟
(1湖南农业大学生物科学技术学院,长沙410128;2湖南杂交水稻研究中心杂交水稻国家重点实验室,长沙410125)
0 引言
【研究意义】水稻的易落粒性状不仅造成稻谷收获产量的损失,而且不适于机械化收割。某些水稻品种(组合)因落粒性强导致收获产量损失程度高达5.8%—8.6%[1],且因水稻机械化生产方式的普及,使易落粒品种在实际生产过程中受到更严重的产量损失;因此,加强对水稻品种落粒性的遗传改良对于保证水稻稳产和适于机械化生产均具有重要意义。【前人研究进展】基因编辑是一项以核酸酶作为基础工具,对生物体基因组中的特定基因进行靶向“修饰”,从而创制预期目标性状的定向遗传改良技术。目前常见的基因编辑技术主要有锌指核酸酶(zinc-finger nucleases,ZFNs)[2]、转录激活子样效应物核酸酶(transcription activator-like effector nucleases,TALENs)[3]和CRISPR/Cas核酸酶(规律成簇间隔短回文重复序列,clustered regularly interspaced short palindromic repeats/CRISPR-associated nucleases)[4]等为工具的3种技术体系,作为第三代基因编辑技术—CRISPR/Cas是一类来源于细菌和古细菌体内的获得性免疫防御系统[5-7],与 ZFNs、TALENs技术相比具有更加简单、高效等优势。2012年JINEK等[8]研究发现TypeⅡCRISPR/Cas系统的Cas9核酸酶结合crRNA和tracrRNA 2个RNA分子就可以切割双链DNA,奠定了 CRISPR/Cas9技术应用的理论基础;2013年ZHANG 等[6]和 MALI等[9]分别在《Science》杂志上发表了2篇基于CRISPR/Cas9技术改造人类与小鼠细胞系基因组的文章,引领了该技术的发展。近年来,CRISPR/Cas9技术已被成功应用于动物[10]、植物[11-13]和微生物[14]的基因组定点编辑。在水稻遗传改良中,该技术已成功实现了对抗性、产量、品质和育性等相关基因的定点编辑,如XU等[15]利用CRISPR/Cas9技术对抗除草剂基因BEL为靶点进行定点突变,成功获得对除草剂-苯达松敏感的突变体水稻材料;WANG等[11]对OsERF922进行定向编辑,提高了受体水稻材料对稻瘟病的抗性;ZHOU等[12]以水稻温敏不育基因TMS5为靶点,成功培育了水稻工程温敏不育系;ZONG等[13]等利用CRISPR/Cas9技术成功构建了水稻基因组定点替换体系。这些成功案例的报道,使得CRISPR/Cas9基因编辑技术在水稻定向遗传改良中的应用越来越广泛。目前,多个控制水稻落粒性的主效基因/QTL已被定位在水稻基因组中不同的染色体上[16-21]。qSH1是控制水稻落粒性的主效基因之一,其在小穗基部的离层持续表达促进了离层的形成,导致水稻籽粒易脱落。研究进一步发现,在qSH1开放阅读框 12 kb处 5′端调控区存在一个能够影响 ABI3型转录因子与RY重复序列结合的SNP变异(G/T),该位点以基因型T存在时能阻碍qSH1的表达,从而抑制离层形成及产生难落粒表型;反之,当其基因型为G时,能维持qSH1的持续表达促进落粒。利用该SNP(TT)对易落粒品种Kasalath中的qSH1基因型进行替换后,因近等基因系中qSH1的表达得到了明显抑制而表现出了难落粒的表型[22]。王军等[23-24]通过对江苏、黑龙江、广东等地区的39份落粒性极强的杂草稻的qSH1等落粒性主效基因的序列分析,证明qSH1是控制这些地区杂草稻极易落粒表型的主效基因且该SNP位点都表现出一致的“GG”基因型。此外,研究发现qSH1对其他落粒性控制位点间存在上位效应和互作关系。ONISHI等[25]研究来自23份不同水稻资源的3个水稻落粒性基因(qSH1、qSH3和qSH4即Sh4),发现在qSH3和qSH4存在时,qSH1的突变可显著地把易落粒表型转变为难落粒,由此说明qSH1对其余 2个水稻落粒性基因具有明显的上位效应。ZHOU等[21]通过原位杂交解析3个与水稻离层发育相关的主效基因(qSH1、Sh4和SHAT1)的遗传关系,发现qSH1在幼穗分化后期开始表达,并作用于Sh4、SHAT1的下游,通过维持其稳定表达,从而促进小穗离层结构的形成。【本研究切入点】前人对水稻落粒性基因/QTL的定位及关联分析表明,qSH1是控制水稻落粒性的关键主效基因,而传统的遗传改良的方法存在效率低、耗地多及不定向等不足。利用基因编辑技术对水稻落粒性主效基因qSH1进行定点编辑,可从分子遗传学层面定向、高效地改良水稻易落粒性这一复杂农艺性状。【拟解决的关键问题】本研究选取了一个综合性状优良但易落粒的大穗型杂交稻骨干亲本 HR1128为受体材料,以落粒性基因qSH1为靶点,利用CRISPR/Cas9技术进行定点编辑,定向改良其落粒性过强的不足。以期为保证水稻稳产和机械化生产创制重要的材料基础。
1 材料与方法
1.1 试验材料
以HR1128为转化受体亲本,2014—2017开展常规的转基因试验,转基因水稻材料种植于湖南杂交水稻研究中心长沙转基因试验基地,常规田间种植及水肥管理。
pYLgRNA-U3/U6a载体及pCRISPR/Cas9表达载体由华南农业大学刘耀光院士惠赠,试验所用引物(电子附表 1)由生工生物工程(上海)股份有限公司合成,测序由湖南擎科生物技术有限公司完成,BsaⅠ、T4 DNA ligase、KOD FX分别购置于NEB、宝生物工程(大连)有限公司、东洋纺(上海)生物科技有限公司,其余分子试剂均购置于天根生化科技(北京)有限公司。
1.2 靶点设计与CRISPR/Cas9表达载体的构建
理论上,在基因的编码区域或者5′-UTR区域进行编辑可能改变基因翻译后的蛋白产物结构或影响该基因的表达水平,是失活基因功能或抑制基因表达的2种基本策略。根据CRISPR/Cas9系统识别原型间隔序列毗邻基序(protospacer adjacent motif,PAM)上游约20个核苷酸序列的特点,在水稻落粒性基因qSH1(LOC_Os01g62920)上设计了 2个靶位点,分别命名为 qSH1-T1和 qSH1-T5(图 1-A)(qSH1-T1 序列 5′-GCGCCATGTCGTCCGCCGCT-3′,PAM 序列为 5′-GGG-3′,qSH1-T5 序列 5′-ACATGGC GCGCACGCACGTA-3′(qSH1反向互补链),PAM序列为5′-CGG-3′)。2个靶点的主要靶标区域分别在qSH1第一个外显子区域和 5′-UTR区域,以便获得qSH1生物学功能丧失或表达量降低的突变体后代。靶点的特异性通过信息网站Cas-OFFinder和NCBI中水稻基因组BLAST对比分析验证,发现2个靶点不会或不易引起脱靶效应。
参照 MA等[26]的方法构建双靶点表达载体pYLCRISPR/Cas9- qSH1-T51。(1)构建含qSH1-T5和qSH1-T1双靶点的pYLgRNA-U3、pYLgRNA-U6a载体:分别合成带有黏性末端的寡链靶点引物 qSH1-T5F/qSH1-T5R和qSH1-T1F/qSH1-T1R。将2对寡链靶点引物变性后冷却至室温完成退火,然后将退火后的2个片段分别接连到pYLgRNA-U3和pYLgRNAU6a载体上,并利用PCR分别扩增获得含有qSH1-T5和qSH1-T1靶点的gDNA表达盒U3-qSH1T5-gRNA和U6a-qSH1T1-gRNA;(2)构建含qSH1-T5和qSH1-T1靶点片段的pYLCRISPR/Cas9表达载体:用接头引物b1/B2和 b2/BL分别对 U3-qSH1T5-gRNA和 U6aqSH1T1-gRNA表达盒进行PCR扩增,再用BsaⅠ酶、T4 DNA ligase将所有扩增产物与pYLCRISPR/ Cas9-MT表达载体同时进行酶切和连接,构建pYLCRISPR/Cas9-qSH1-T51表达载体。(3)转化及测序验证:将连接产物转化至DH5α感受态细胞中,挑取单菌落扩繁培养,抽提质粒DNA后用AscⅠ酶切鉴定检测,并挑选AscⅠ酶切正确的质粒DNA用检测引物SP1/SP2(电子附表1)进行PCR扩增,回收扩增产物送湖南擎科生物技术有限公司测序。将通过酶切和测序验证正确的表达载体转化农杆菌感受态EHA105。
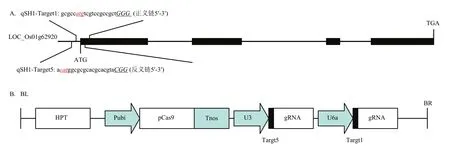
图1 gRNA靶位点及pYLCRISPR/Cas9-qSH1-T51表达载体组装示意图Fig. 1 Target sites of the gRNA in the qSH1 gene and construction of the pYLCRISPR/Cas9-qSH1-T51 vector
1.3 转基因阳性植株的获得
使用构建好的阳性农杆菌株转化水稻品种HR1128的愈伤组织,用潮霉素筛选得到的再生组培苗,即为T0代植株(水稻遗传转化委托武汉伯远生物科技有限公司完成)。采用 CTAB法[27]提取 T0代植株的基因组 DNA,用潮霉素基因特异引物 hpt-F1/hpt-R1(电子附表1)进行PCR扩增,能扩增出目的片段的植株即为T0代转基因阳性植株。
1.4 靶位点检测
为检测靶位点的突变情况,跨 qSH1-T1和qSH1-T5 2个靶点设计靶点检测引物 qSH1-JC-F1/qSH1-JC-R1(电子附表1),扩增产物片段大小约为671 bp。用qSH1-JC-F1/qSH1-JC-R1对T0代转基因阳性株进行PCR扩增,将扩增产物送湖南擎科生物技术有限公司测序。利用 MAGE4.0分析测序结果,在靶点位置出现双峰、套峰的植株即为T0代突变植株。再将突变植株的扩增产物进行TA克隆,随机挑选10个单克隆测序,测序结果与野生型序列进行对比以分析突变类型及基因型。
1.5 无T-DNA元件的qsh1突变系的获得
筛选得到的 T0代突变株自交结实后得到 T1代种子,将 T1代种子播种成苗移栽后提取叶片基因组DNA,用hpt-F1/ hpt-R1引物(电子附表1)进行PCR扩增,扩增不出目的条带的植株即为无 T-DNA成分的T1代单株。再对上述单株用qSH1-JC-F1/ qSH1-JCR1(电子附表1)引物进行靶点PCR扩增并测序分析,筛选纯合突变单株即为T1代无T-DNA成分的qsh1纯合突变体。对上述筛选的qsh1纯合突变体自交加代,获得T2代qsh1突变系。
1.6 潜在脱靶位点分析
利用NCBI、Gramene的BLAST工具选取Oryza sativa IndicaGroup为参考序列,筛选与每个sgRNA序列匹配度大于或等于15 bp且3′端具有NGG的位点作为潜在脱靶位点,评估各个靶位点的脱靶效应。每个潜在脱靶位点分别检测T0代、T1代转基因阳性植株。
1.7 落粒性测定分析
采用随机分区设计种植无T-DNA成分的qsh1突变系及HR1128野生型对照,3个重复,每个小区中各株系种5行,每行8株。每个株系黄熟后取中间5株的主穗测定其穗尖籽粒的脱落拉力,每穗各测定10粒,每个株系设置3个重复。脱落拉力采用拉力计测定,各株系测定的拉力值采用SPSS13.0计算平均值和标准差,并利用单因素方差分析法比较不同株系间拉力值的差异,显著性差异水平为P≤0.05,计算得出的平均拉力值与该株系的落粒性呈负相关性。
1.8 qsh1突变系主要穗部农艺性状考查与分析
于成熟期,选择qsh1突变系和HR1128野生型材料的主穗,考查每穗总粒数、结实率并随机称取250粒饱满种子的质量换算成千粒重,每个株系各考查5个单株。获得的数据采用SPSS13.0进行统计分析。
1.9 qsh1突变系qSH1相对表达量分析及编码氨基酸序列预测分析
提取黄熟期qsh1突变系和HR1128野生型穗部总RNA,并反转录合成第1链cDNA。利用实时荧光定量PCR(qRT-PCR)方法分析qSH1在突变体和野生型中的表达水平,其中以qSH1-Q-F/qSH1-Q-R目的基因引物(扩增区段位于qSH1第 2个外显子区)、UbQ5-Q-F/UbQ5-Q-R为内参基因引物,利用2-ΔΔCT方法计算基因相对表达量。同时,采用 MAGE4.0和DNAMAN对突变体和野生型qSH1编码的第一个外显子氨基酸序列进行进一步的预测比对分析。
2 结果
2.1 qSH1靶点设计和pYLCRISPR/Cas9-qSH1-T51表达载体构建
根据 CRISPR/Cas9技术的原理并结合qSH1的cDNA序列,在起始密码子附近找到2条长度为20 bp特异性好的片段,将其设计为 2个靶点 qSH1-T5和qSH1-T1(图 1-A)。然后利用酶切连接、PCR等方法将qSH1-T5、qSH1-T1与gRNA表达盒连接,最后再将 2个连接成功的靶点-gRNA同时组装至pYLCRISPR/Cas9-MT表达载体(图1-B)。组装好的表达载体 2个靶点-gRNA表达盒分别由OsU3和OsU6a启动子驱动,Cas9由Ubi启动子驱动。
2.2 T0代转基因阳性苗的获得及qSH1编辑情况分析
用含有 pYLCRISPR/Cas9-qSH1-T51表达载体的农杆菌转化HR1128,获得T0代再生苗。载体特异引物 hpt-F1/hpt-R1(电子附表 1)检测结果表明,获得的20株T0代再生苗中有11株为转基因阳性苗,转基因阳性率为55%。再对获得的转基因阳性苗用靶点检测引物qSH1-JC-F1/ qSH1-JC-R1(电子附表1)对qSH1靶点上下游区域进行PCR扩增,并将扩增产物进行测序分析,结果表明,有7株转基因阳性苗在qSH1靶点位置发生了编辑(图2),其中T1靶点的突变频率为54.55%、T5靶点的突变频率为63.64%、T1和T5靶点同时突变的频率为 54.55%(T1靶点发生突变的单株,其在T5靶点都发生了突变)。
进一步对7个突变单株靶点扩增产物进行TA克隆,并随机挑选10个单克隆测序分析,结果表明,qSH1的突变基因型包括纯合突变、杂合突变、双等位突变和嵌合突变,突变类型包括碱基插入、碱基缺失及碱基替换(表1)。
2.3 无T-DNA成分的qsh1突变系的获得
为获得不含 T-DNA成分且突变位点能稳定遗传的qsh1突变系,将7个T0代qsh1突变单株自交繁殖,构建7个T1代qsh1突变群体。利用hpt-F1/hpt-R1和qSH1-JC-F1/qSH1-JC-R1引物(电子附表1)对T1代突变群体中单株的载体序列和靶点区域进行扩增测序分析。发现在T1代植株中Cas9载体骨架和qSH1突变位点都发生了分离(图 3),并筛选获得 2种突变类型不同且无T-DNA成分的qsh1纯合突变。将上述2种纯合突变株自交加代,进一步构建2个T2代qsh1纯合突变系群体,命名为17SZ01和17SZ02(图4)。
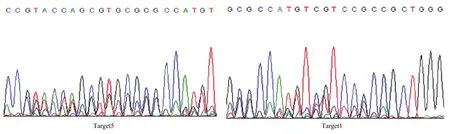
图2 2个靶位点的测序结果Fig. 2 Sequencing result for the two target sequences
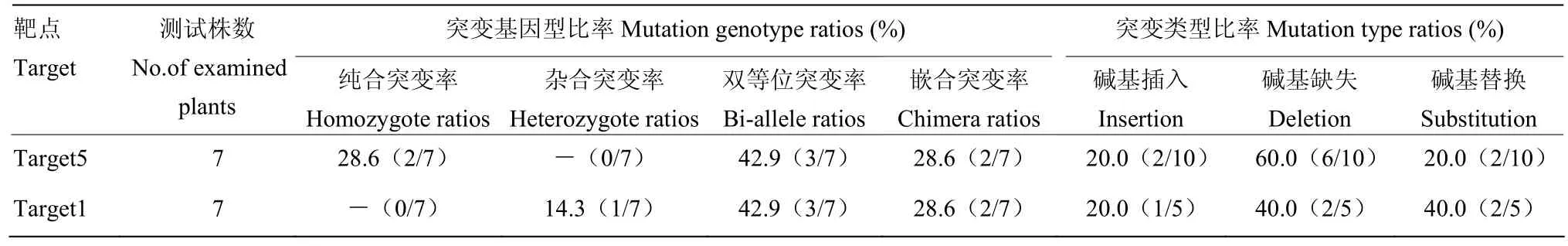
表1 T0代突变体突变基因型比率、突变类型比率Table 1 The genotype ratios and mutation type ratios of T0 plants
2.4 CRISPR/Cas9系统潜在脱靶效应的分析
根据靶位点 qSH1-T5、qSH1-T1序列与Oryza sativa IndicaGroup参考序列的比对结果,筛选出的3个潜在脱靶位点。并对获得的 T0和 T1代转基因阳性植株46株进行潜在脱靶位点PCR扩增测序,结果比对分析发现,所有被检测植株的潜在脱靶位点均没有发生突变(表2)。
2.5 qSH1编辑后代落粒性的测定及主要穗部农艺性状的考查分析
于成熟期,采用拉力计对qsh1纯合突变系17SZ01、17SZ02及HR1128野生型对照单株主穗或大分蘖穗尖部的籽粒脱落拉力进行测定分析。结果表明,所测定的T2代qsh1纯合突变系的拉力值与野生型对照相比显著增加(拉力值分别为(116.38±15.48)g和(103.73±15.70)g),由此说明qsh1纯合突变系的落粒性呈现显著性降低(图5)。同时,对qsh1突变系、HR1128野生型材料的每穗总粒数、结实率、千粒重等几项主要穗部农艺性状进行考查统计分析发现,各项指标在突变系和野生型材料间均无显著性差异(表3)。

图3 部分qsh1突变株T-DNA成分的PCR鉴定Fig. 3 PCR identification for the T-DNA elements of parts of the qsh1 mutants
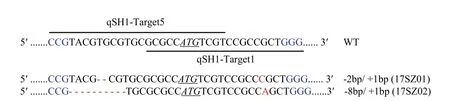
图4 T2代2种不同类型qsh1纯合突变的突变类型分析Fig. 4 Mutation types of two types qsh1 homozygotes in T2 generation
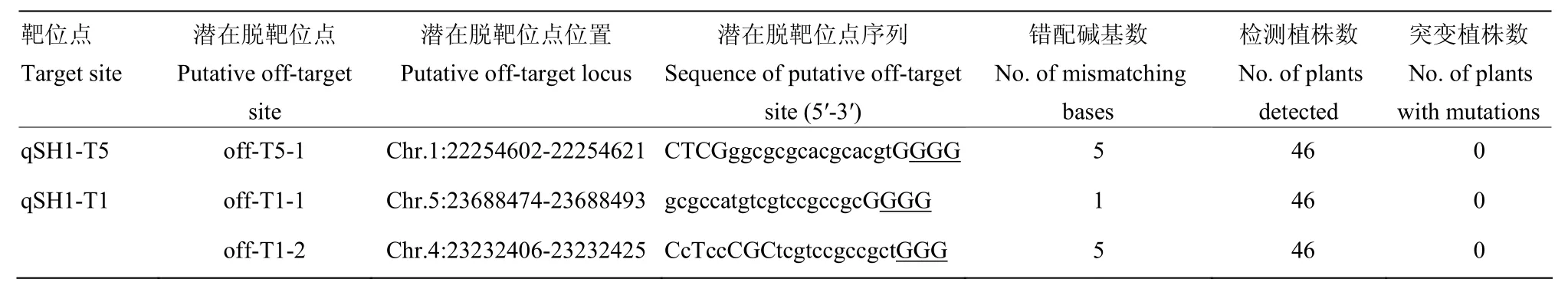
表2 CRISPR/Cas9-qSH1-T51潜在脱靶位点的检测Table 2 Mutation detections in the putative CRISPR/Cas9-qSH1-T51 off-target sites

图5 T2代qsh1纯合突变系及HR1128野生型落粒性分析Fig. 5 The seed shattering of qsh1and HR1128 in T2 generation
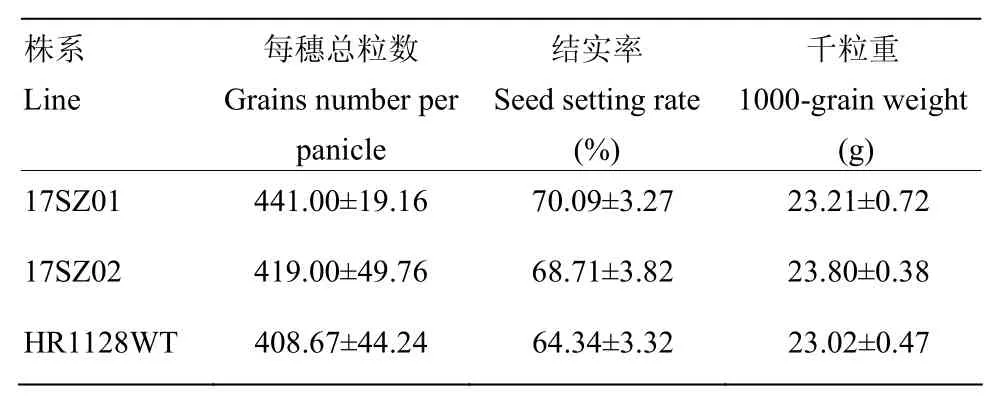
表3 qsh1突变系和HR1128野生型主要穗部农艺性状调查Table 3 Major panicle agronomic traits of qsh1 mutants and HR1128 WT
2.6 突变系qSH1的qRT-PCR检测及氨基酸序列预测分析
对qsh1纯合突变系17SZ01、17SZ02及HR1128野生型对照黄熟期穗部qSH1进行qRT-PCR检测并对其相对表达量进行计算分析,发现与野生型对照相比,17SZ01突变系qSH1的相对表达量显著降低(图6),而17SZ02突变系qSH1的相对表达量无显著差异。
同时对2个qsh1纯合突变系编码蛋白的氨基酸序列进行预测比对,发现17SZ01和17SZ02均由于靶点T1的插入突变引发qSH1第一个外显子编码区移码,从而导致氨基酸翻译发生改变并提前终止(图7)。
3 讨论
CRISPR/Cas9技术是目前最前沿的基因编辑技术,相对于锌指蛋白(zinc-finger nucleases,ZFNs)和TALE核酸酶(transcription activator like effector nucleases,TALENs)技术,其具有载体构建简便、靶位点选择广泛及打靶效率高等优势,目前已成功用于水稻[11-12,15,28]、玉米[29]、小麦[30]等多种作物特定基因的定点编辑且多用于受体材料的定向遗传改良。
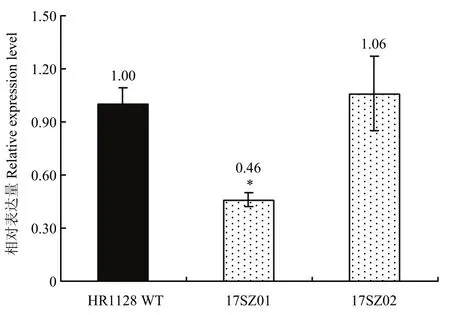
图6 qsh1纯合突变系及HR1128野生型qSH1相对表达量Fig. 6 qSH1 relatively expression level of qsh1 mutant line and HR1128 WT
本研究利用双靶点CRISPR/Cas9表达载体对水稻的qSH1进行定点编辑,成功获得一系列不同突变类型的qsh1突变体。T0代qsh12个靶点的突变频率分别为 54.55%和 63.64%,突变基因型以双等位突变频率最高(T5、T1均为42.9%),突变类型以10 bp以内小片段的碱基缺失为主(T5和T1分别为60.0%和40.0%),这与前人研究结果一致[31-32]。对T1代单株的靶位点和T-DNA元件的检测发现在T1代植株中突变位点和T-DNA载体骨架都发生了分离,由此从T1分离群体中筛选得到不含T-DNA成分的qsh1纯合突变体,并进一步自交获得T2代qsh1突变系。同时,对 T0和 T1代转基因阳性植株的潜在脱靶位点进行了脱靶效应的检测分析,发现被检测的植株均未发生脱靶现象。
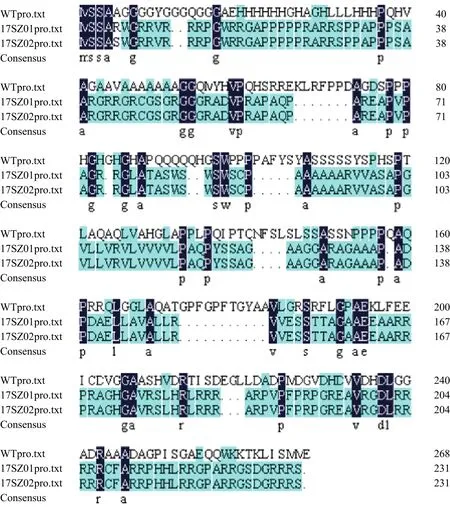
图7 qsh1纯合突变系及HR1128野生型 qSH1第一个外显子预测氨基酸序列分析Fig. 7 Analysis of qSH1 gene the first extron putative amino acid sequences of qsh1 mutant lines and HR1128 WT
通过对黄熟后的T2代qsh1纯合突变系籽粒脱落拉力的测定分析,与野生型对照相比,2个突变系落粒性有显著性降低(籽粒脱落拉力值分别为(116.38±15.48)g和(103.73±15.70)g)。发现 17SZ01和 17SZ02突变系均在qSH1的第一个外显子上发生了碱基插入,使该基因编码区产生移码突变,从而导致其编码的氨基酸改变且qSH1蛋白翻译提前终止,最终引起该蛋白不能发挥正常的生物学功能,可能是造成突变系落粒性显著降低的主要原因。而 17SZ01突变系的qSH1表达量发生显著下降,是否对突变系落粒性表型的改变起到增效作用?后续仍需更多不同突变类型的纯合系进行分析,以验证推论是否正确。据报道,5′-UTR作为连接第一个外显子和启动子的非编码区,该区域在基因的转录和翻译过程中起着重要的调控作用,该区域的突变可直接影响启动子活性,进而影响下游基因表达[33-35],所以本研究分析认为17SZ01突变系在qSH1的5′-UTR区的缺失突变,可能引起了其调控位点的功能变化或丧失,影响了该基因启动子的活性,从而导致了该突变系qSH1的表达量显著下降。同时,由于第一个外显子上的插入突变可能激活了植物体细胞内无义介导的mRNA降解(nonsense-mediated mRNA decay,NMD)途径[36]及转基因组培操作过程中可能诱发的转座子激活等现象[37],都可能影响mRNA的稳定性,从而造成突变系基因表达的变化。
传统的遗传改良育种技术周期长、效率低,且容易带入连锁累赘基因。CRISPR/Cas9基因编辑技术可以快速、高效地定点编辑特定基因定向改良受体材料的目标性状,而且在自交后代中分离得到无 T-DNA成分的纯合突变株系,极大地缩短了水稻遗传改良育种的周期。本研究定点编辑了易落粒的大穗型籼稻品种HR1128中qSH1,并在得到的突变体后代中获得了落粒性状显著降低的改良株系。目前,笔者正在将改良的突变系材料与野生型亲本进行回交选育,进一步纯化突变系后代的遗传背景。并同时筛选更多不同突变类型的纯合系,解析不同位点突变对降低落粒性的改良效果以挖掘最适的突变位点,为定向改良某些水稻品种(组合)易落粒这一复杂农艺性状探索了一条全新、绿色、高效的分子育种途径。
4 结论
利用 CRISPR/Cas9技术对易落粒的杂交稻亲本品种HR1128的qSH1进行定点编辑,创制了一批qsh1突变体材料,并筛选获得落粒性显著降低的改良后代。这是一条全新、绿色、高效的分子育种策略。
致谢:pYLgRNA-U3/U6a载体及pCRISPR/Cas9表达载体由华南农业大学刘耀光院士提供;文章的撰写及修改得到彭彦博士、潘银林博士的帮助,在此表示感谢。
