1-脱氧野尻霉素与糖代谢酶相互作用的分子动力学计算
2018-07-11李林周虹唐忠海苏小军李清明郭时印
李林,周虹,唐忠海,苏小军,3,李清明,郭时印
(1.湖南农业大学食品科技学院,湖南长沙 410128)(2.湖南农业科学院作物研究所,湖南长沙 410125)(3.湖南省作物种质创新与资源利用重点实验室,湖南长沙 410128)
现有研究报道,1-脱氧野尻霉素(DNJ)由于其结构与葡萄糖相似,可竞争性抑制糖苷酶活性,从而显著降低多糖的降解水平,现有关于DNJ的研究集中分离提取工艺的优化、提取物生物活性和物理化学性质的研究[1]。DNJ同时与糖尿病相关的五种酶作用的机制尚未见报道,在相互作用机制没得到合理阐述之前,传统的体外筛选试验往往具有很大盲目性与随机性,成本较高,也将影响DNJ在食品或药品中的开发及利用。
分子对接是将配体分子置于受体分子的活性位点附近,搜索分子自身的构象和位置,得到与分子间相互作用的最佳匹配模式。目前已经广泛应用于小分子与蛋白结合模拟方面,在候选药物活性化合物的发现及其与靶标的结合方式方面已经得到认可[2]。分子动力学模拟方法是一种具有确定性的模拟方法,其通过求解一组原子运动方程得到整个体系的相空间运动轨迹,再利用统计学方法得到其宏观性质。分子对接是从整体上考虑配体小分子与受体大分子(生物大分子)的结合情况,在一定程度上能有效地防止不同方式里部分作用稍好但整体效果不完善这种高频出现的尴尬现象。其科学性及确定性越来越受到科研工作者的青睐[3]。另外有很多研究者感兴趣但目前的条件无法进行的实验也能通过计算得以完成,比如分子动力学模拟可以在纳秒级对分子行为进行分析。
但是分子对接这项技术的缺陷也十分明显,在分子对接体系中,我们在很多情况下默认小分子和生物大分子进行的是刚性对接即不考虑分子的振动转动等因素对配体与受体结合的影响,而实际环境中小分子配体和生物大分子都是处于一个不断变化的过程中,所以还需要借助分子动力学更好地模拟小分子配体和生物大分子的最真实的模拟情况。基于此,本试验采用分子对接和分子动力学模拟方法,从 PTP1B、PPARα、PPARγ、α-淀粉酶(α-Amylase)和α-葡萄糖苷酶(α-Glucosidase)等蛋白中确定了DNJ的作用靶标,并阐明其作用模式。
1 计算方法
1.1 分子对接
分子对接的准备工作均在 UCSF Chimera[4]中进行。首先,从RCSB Protein Data Bank上下载PTP1B、PPARα、PPARγ、α-淀粉酶和α-葡萄糖苷酶的晶体结构(PDB编号:4Y14、5HYK、3U9Q、4GQR和3WY2,分辨率:1.90Å、1.83Å、1.52Å、1.20Å和1.47Å)。去除蛋白晶体中多余肽链和溶剂小分子,除PPARγ保留A、B两链外,其他蛋白均保留A链。配体分子1-脱氧野尻霉素的三维结构下载自PubChem。然后,采用DockPrep模块为蛋白添加氢原子,为蛋白和配体分子分别添加AMBERff4SB力场和AM1-BCC电荷[5,6],并对配体分子进行了充分的能量优化。接着,采用DMS工具以半径为 1.4Å的探针生成受体的分子表面,以晶体中的配体化合物所在位置为对接口袋,使用 sphgen模块生成围绕活性位点的球状集合(Spheres),选择距离活性位点中心8Å以内的球集,包围球集的盒子大小设为12Å×12Å×12Å。使用DOCK 6.7[7,8]软件包的Grid程序生成用于能量打分的Grid文件。在本对接研究中,采用半柔性对接方法(即配体分子的构象可发生变化,蛋白受体的结构保持刚性),为各体系分别生成10000个不同的分子构象取向,计算配体分子与结合位点的静电力和范德华力贡献,加和得到Grid打分。经过聚类分析(RMSD截断值为2.0Å,避免生成冗长多余构象),得到若干打分最佳的对接构象。采用人工分析方法,从各体系中选取一个最佳构象用于分子动力学模拟。
1.2 分子动力学模拟
选取各体系打分最佳的构象(构象 1)作为分子动力学模拟的初始结构,所有准备和模拟工作均在Amber14和AmberTools15[9]中进行。采用tLeap工具为受体蛋白和配体小分子分别添加AMBERff14SB和GAFF力场,小分子采用AM1-BCC方法计算的电荷。然后,体系采用TIP3P水模型,水箱大小为10Å,添加抗衡离子(counter ions)使体系接近电中性。经过以下步骤使体系达到平衡:(1)2000步能量优化:复合物原子添加约束力100 kcal/(mol·Å2),其中前面800步采用最陡下降法,然后共轭梯度法进行之后 1200步的优化;(2)3000步能量优化:受体重原子添加约束力50 kcal/(mol·Å2),前面1000步采用最陡下降法,接着2000步采用共轭梯度法;(3)8000步能量优化:不加约束力,前面2000步最陡下降法,接着6000步采用共轭梯度法;(4)50 ps的加热相:受体和配体全部原子添加约束力100 kcal/(mol·Å2),最终温度为298 K;(5)50 ps平衡相:受体和配体全部原子添加约束力50 kcal/(mol·Å2),采用NPT系综(isothermal is obaric ensemble);(6)100 ps平衡相:受体和配体全部原子加约束力50 kcal/(mol·Å2),采用NPT系综。但考虑到计算代价,对各个体系仅进行1ns的NPT系综动力学模拟,采用朗之万动力学(Langevin dynamics)方法控制体系温度,设定碰撞频率(collision frequency)为10 ps-1,所有形成共价键的氢原子采用SHAKE[10]算法加以约束,远程静电相互作用(long range electrostatic interactions)采用 Particle Mesh Ewald(PME)[11]方法进行处理,范德华相互作用(vander Waals interactions)截断值设为10Å。采用MM/GBSA[12]方法计算结合自由能。
出于计算资源成本与计算结果精度的考虑,首先采用Amber14为各体系进行1ns的分子动力学模拟,通过结合自由能计算,确定DNJ的潜在作用靶标,然后,对该体系进行20 ns的分子动力学模拟,利用氢键分析和能量分解手段,阐明其相互作用模式及降血糖作用的分子机制。
2 计算结果
2.1 分子对接结果
采用DOCK6.7预测配体小分子DNJ与PTP1B、PPARα、PPARγ、α-淀粉酶(α-Amylase)和α-葡萄糖苷酶(α-Glucosidase)等蛋白的结合模式,经过聚类计算,各自得到2~4个对接构象,打分情况如表1所示

表1 配体小分子DNJ与各受体蛋白的对接打分(kcal/mol)Table 1 Scores of DNJ docking with the proteins
对接构象的Grid Score总打分在-41~-30 kcal/mol之间,尽管静电力贡献 Grid_es差异相对较大,但同一体系的对接构象之间的范德华力贡献 Grid_vdw差别不大,因而构象间Grid Score总打分差别不大。这是因为小分子DNJ结构简单且体积小,可变构象少,导致它与各蛋白的接触面积较小,疏水作用不明显。经分析,在各蛋白口袋中,DNJ各对接构象的取向和结合方式较为接近,因此,选取打分最佳的对接构象(构象1)用于后续研究。
2.2 分子动力学模拟
2.2.1 RMSD分析
为了解体系动力学过程的稳定性以及结合自由能取样的合理性,使用 Cpptraj模块对动力学骨架进行RMSD分析。体系动力学平衡阶段的第一个构作为RMSD的参照构象,只考虑蛋白的骨架原子(CA、C和N原子)和配体分子的重原子。
RMSD的变化显示了体系整体结构的波动情况。如表所示,各体系整体和蛋白自身在经历短暂的时间后迅速达到平稳,DNJ小分子的RMSD在PPARα-DNJ体系中500 ps附近出现了跳跃,然后立即进入新的平台。检查结构发现,该微小的跳跃(小于0.5 Å)是由于DNJ小分子轻微的构象调整,由于形成新的氢键而立即稳定下来。因此,各体系已达到平衡状态,可进行后续分析。
2.2.2 结合自由能计算
Amber14中结合自由能是通过对分子动力学轨迹进行定点采样统计得到的平均值。
在本研究中,对整个平衡阶段1 ns的动力学轨迹进行采样,选取200帧,采用MM/GBSA方法进行结合自由能计算。出于计算成本的考虑,结合自由能计算中并未考虑熵的贡献。然而,由于DNJ小分子为一小环结构,结合前后自身构象变化不大,蛋白受体也未见发生较明显的蛋白运动,因此,熵对结合力计算影响不大。
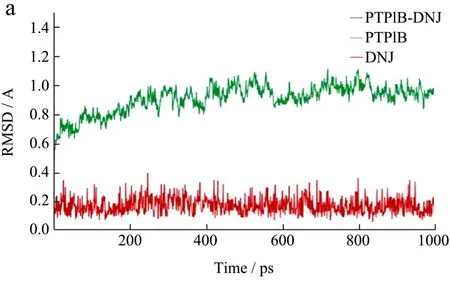
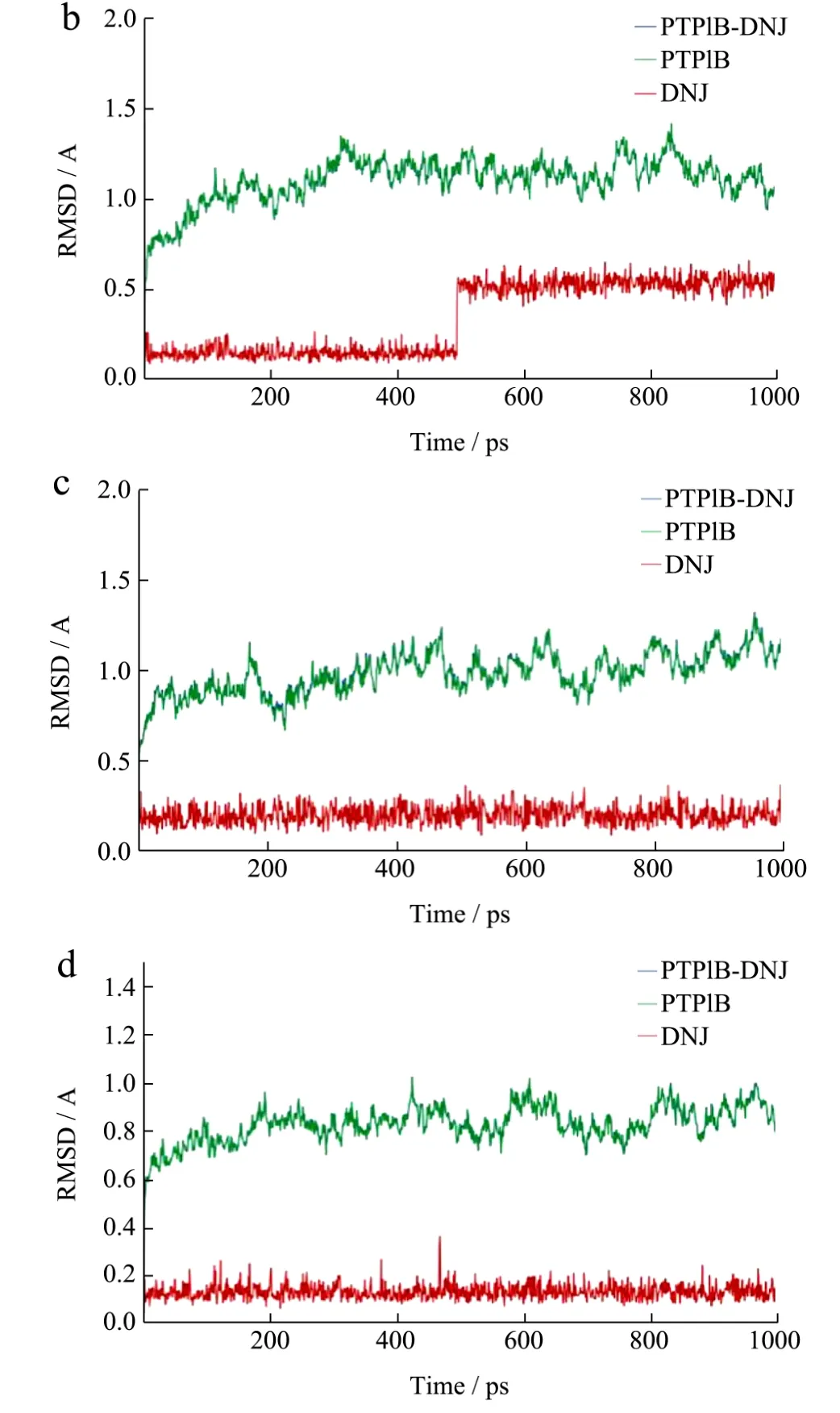

图1 各体系1ns分子动力学轨迹的RMSD分析Fig.1 RMSD analysis of 1ns molecular dynamic track of every systerm
如图1所示,PTP1B、PPARα、PPAPγ、α-Amylase和α-Glucosidase这5个体系的结合自由能计算值分别为-24.66 kcal/mol、-25.04 kcal/mol、-21.65 kcal/mol、-21.84 kcal/mol和-35.76 kcal/mol。DNJ小分子与α-Glucosidase的结合力显著大于其他体系,提示DNJ小分子极有可能作用于α-Glucosidase。而自由能较小的其他蛋白,则可能不是DNJ小分子的靶标。分析能量项可知,在形成复合物的过程中,范德华力作用、静电力作用和非极性溶剂化能均为负值,有利于复合物结合;而极性溶剂化能则为正值,对复合物的形成有阻碍作用。α-Glucosidase复合物的范德华力贡献在5个体系中偏低,但静电力贡献很好地弥补了这一不足。因此,静电力作用是DNJ小分子与α-Glucosidase蛋白结合的主要驱动因素,这与其小体积的极性结构特点相一致。

表1 各体系的结合自由能(kcal/mol)Table 2 Free Combined energy of every systerm (kcal/mol)
2.2.3 能量分解

图2 α-Glucosidase-DNJ体系20 ns分子动力学轨迹的RMSD分析Fig.2 RMSD analysis of 20 ns molecular dynamic track of 2α-glucosidase-DNJ systerm

图3 α-Glucosidase与DNJ小分子结合的关键相互作用Fig.3 Important interactions between α-glucosidase and DNJ

图4 α-Glucosidase与DNJ小分子的结合模式Fig.4 Combining mode of α-glucosidase with DNJ
根据结合自由能计算结果,推测α-Glucosidase是DNJ小分子的靶标。为深刻理解它们的相互作用模式以及更具体地分析作用的选择性问题,对该体系增加20 ns的动力学模拟,并对体系进行能量分解,采用MM/GBSA方法计算所有残基的贡献。
为考察体系在长时间模拟中是否发生较大的构象变化,进行了RMSD分析。结果表明(图2),蛋白骨架原子经过约3 ns的运动调整后,达到长时间的平衡。DNJ分子在3 ns时发生了较明显的构象变化,并维持至10 ns,但之后恢复到原来的构象,并最终达到平衡,一直维持到模拟结束。因此,截取10 ns后的轨迹进行聚类分析、氢键分析和结合自由能相关计算。
能量分解结果表明(图3 ),Glu271、Asp333和Asp202在DNJ与α-Glucosidase结合中作用明显。其他残基,如His332、Asn331也对复合物的形成做出正向贡献,而Asp62则不利于复合物的形成。对于DNJ和α-Glucosidase的复合物,范德华力和非极性溶剂化能的贡献很小,上述重要残基的结合力贡献主要来自静电力作用和极性溶剂化能,但两者发生一定程度的抵消。
Asp202中的羧基带负电,与 DNJ小分子的 O1羟基之间存在很大的静电吸引力,真空静电作用能为-8.12±1.54 kcal/mol,但极性溶剂化能为 6.91±1.44 kcal/mol;氢键分析表明,两者之间形成一个平均长度2.76 Å,键角159.55°的氢键,占有率达98.14%,非常稳定。Glu271的羧基与DNJ的O2和O3分别形成2个氢键作用,键长 2.68Å 和 2.74Å,键角 164.83°和158.35°,占有率89.22%和105.05%(表明在模拟过程中,羧基发生旋转,存在O3原子同时与羧基的两个氧原子形成氢键的情况)。因此,其静电力贡献最大,达到-15.85 kcal/mol,极性溶剂化能虽然也增大不少,综合而言,总结合力贡献也为最大(-3.86 kcal/mol)。Asp333的羧基O原子同时与DNJ的羟基O原子和N原子形成氢键作用,键长 2.67Å 和 2.866 Å,键角162.38°和161.17°,占有率96.55%和74.05%。
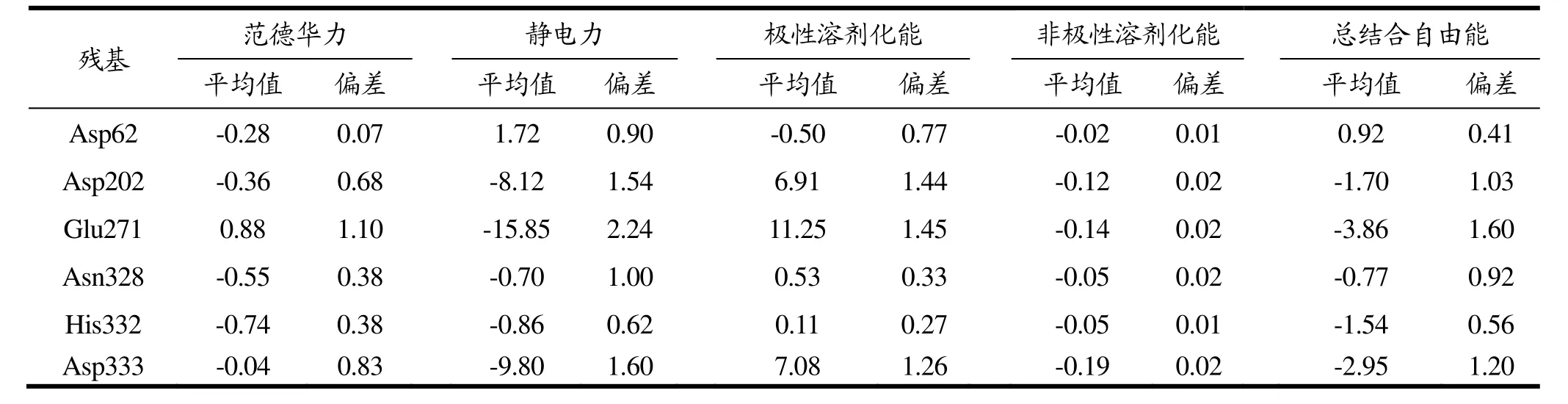
表2 采用MM/GBSA方法进行能量分解的主要贡献残基各部分能量值(kcal/mol)Table 3 The energy value of the main residues analysised with MM/GBSA methord (kcal/mol)
这为DNJ分子的结合提供了-9.80±1.60 kcal/mol的静电力作用,由于极性溶剂化能的抵消作用,总结合自由能贡献为-2.95±1.20 kcal/mol。Asp62的静电力作用为正值,极性溶剂化能抵消不了其排斥作用,因而远离DNJ分子。His332的各项作用力均较小,累计的结合自由能仅-1.54 kcal/mol。其范德华力和静电力均为负值,这提示,结合其带正电的特性可通过结构修饰、添加羧基一类带负电或作为氢键供体的取代基有利于增强DNJ的结合力。图4中DNJ显示为棍棒模型,酶显示为飘带构型,所有关键氨基酸残基显示
为棍棒模型,氢键显示为黄色虚线。

表4 主要残基的氢键分析Table 4 Hydrogen bond analysis of main residues
3 结论

3.1 经过试验,得出五个与糖尿病相关的蛋白质酶分子 PTP1B、PPARα、PPARγ、α-淀粉酶和α-葡萄糖苷酶分别与小分子DNJ的分子对接计算的结果,根据对接打分和结合模式分析,初步认为DNJ小分子的作用
3.2 之后再对5种蛋白分别进行了1 ns的分子动力学模拟,采用MM/GBSA方法计算了各体系的结合自由能。比较发现,α-葡萄糖苷酶与DNJ的结合力有绝对优势。因而,推测α-葡萄糖苷酶是DNJ的一个作用靶标。通过延长模拟时间(20 ns)及引入氢键分析、聚类分析和能量分解等多种分析方法,从分子水平上精确地阐明了DNJ与α-葡萄糖苷酶的相互作用模式(结合模式、作用力类型及大小)与DNJ降血糖作用的分子机制。
3.3 氢键分析和能量分解结果表明,Asp202、Glu271和Asp333是最主要的关键残基,在DNJ与α-葡萄糖苷酶结合中有重要作用,其作用方式是形成稳定的氢键作用,其次为Asn331和His332,通过疏水作用和静电吸引增强DNJ的结合力。由于DNJ分子体积小,范德华力和非极性溶剂化能贡献相对较低,静电力作用和极性溶剂化能是主要的结合力来源。DNJ小分子通过与上述关键残基的作用稳定结合在α-葡萄糖苷酶口袋内部,从而抑制α-葡萄糖苷酶的生物活性,发挥降血糖作用。
