How to Synthesize Vitamin E
2018-07-03WANGZheMAOShanjunLIHaoranWANGYong
WANG Zhe, MAO Shanjun, LI Haoran, WANG Yong
ZJU-NHU United R&D Center, Institute of Catalysis, Department of Chemistry, Zhejiang University, Hangzhou 310028, P. R. China.
1 引言
维生素 E (VE)是生育酚类物质的总称,是一种金黄色或者淡黄色的油状物,带有温和的特殊气味。通常维生素E在光照下遇空气易被氧化而呈现暗红色。它可与丙酮、氯仿、乙醚或者植物油混溶,几乎不溶于水。维生素E是最早发现的维生素之一,由加州大学Evans和Bishop首次发现。随后在 1924年,阿肯色大学 Sure将该物质命名为维生素E。它是一种人体必需的脂溶性维生素,作为一种优良的抗氧化剂和营养剂1,被广泛应用于临床、医药、食品、饲料、保健品和化妆品等行业。
天然存在的维生素 E有四种生育酚(tocopherol)和四种生育三烯酚(tocotrienol)共八种类似物,如图1所示。其中α-生育酚含量最高,生理活性也最高。Evans和Emerson在1936年从麦芽胚中成功分离出结晶状的维生素E,并命名为生育酚。维生素E的首次人工合成发生于1938年,由瑞士科学家Karrer完成。他以三甲基氢醌和植基溴为原料,以无水ZnCl2为催化剂,成功得到了D,L-α-生育酚。同年,Smith、Bergel等人采用异植物醇和三甲基氢醌为原料,以ZnCl2为催化剂合成维生素E,这一方法沿用至今。目前,化学合成的维生素E占总产量的80%以上,而新和成(NHU),巴斯夫(BASF),帝斯曼(DSM)和浙江医药公司是主要的维生素 E供应商,这几家公司生产的产品占据了全球绝大部分的维生素E市场份额。
本综述以 α-生育酚为例,结合本课题组的相关研究内容以及工业化方面的经验,对整个分子的合成过程进行详细的阐述及分析,并对一些可利用的新反应及原料进行展望。α-生育酚的分子结构可以分成两部分,即芳环部分和侧链部分,目前几乎所有的维生素 E工业合成过程都是将三甲基氢醌和异植物醇缩合而成(图2),因此本文将依次介绍如何构筑这两个组成基元和如何将两个基元缩合为 α-生育酚,此外,还会包含一些新颖的合成路线。本文注重合成路线和催化剂的选择,这可以为维生素E的工业合成提供一些新的思路。由于α-生育酚(图1)是一个手性分子,本综述只关注整个分子的构筑,手性合成并不在讨论范围之内。
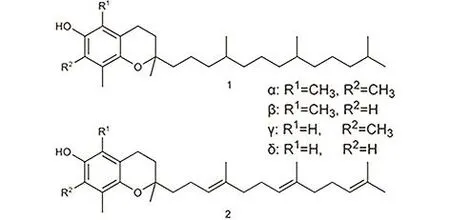
图1 维生素E的结构示意图(1:生育酚,2:生育三烯酚)Fig. 1 Structure of Vitamin E (1 represents tocopherol and 2 denotes tocotrienol).
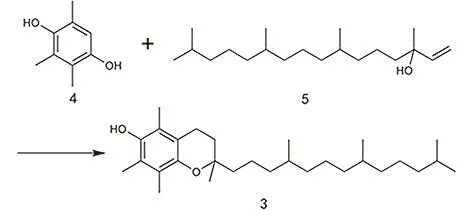
图2 三甲基氢醌(4)与异植物醇(5)缩合生成α-生育酚(3)Fig. 2 The condensation of 2,3,5-trimethylhydroquinone(4) and isophytol (5) to produce α-tocopherol (3).
2 2,3,5-三甲基氢醌的合成
2,3,5-三甲基氢醌(TMHQ),又名2,3,5-三甲基对苯二酚,为白色针状结晶,纯物质熔点在170–173 °C,难溶于水,易溶于醇酮等有机溶剂。TMHQ可以直接用作抗氧化剂。它的合成工艺根据原料可大致分为间甲苯酚法、对叔丁基苯酚法、苯酚法、二乙基铜法、偏三甲苯法、异丙苯偏三甲苯法以及异氟尔酮法。
2.1 间甲苯酚法
间甲苯酚可由煤焦油制取,在一些工业化大国,也可由石油产品制造,来源较为丰富。通常从间甲苯酚出发,加入甲醇发生邻位甲基化反应生成2,3,6-三甲基苯酚,随后氧化为2,3,5-三甲基苯醌(TMBQ),最后加氢为三甲基氢醌(图3)。该工艺流程短,产品收率较高,污染小,是目前三甲基氢醌的主要合成路线2。
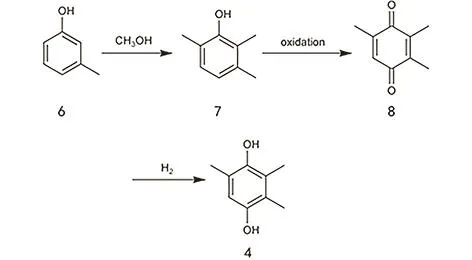
图3 间甲苯酚法制备TMHQ的工艺路线图Fig. 3 The preparation of TMHQ by using m-cresol as a source.
该工艺的关键在于邻位甲基化反应的催化剂,产物选择性尤为重要,除了避免对位或者间位甲基化外,还要避免氧位点的甲基化成醚。最近,Eckstein等3研究了 MFI45催化剂上苯酚的乙基化反应(乙醇为烷基化试剂),发现邻位、对位以及O位的乙基化活化能差别很小,都约为 104 kJ·mol−1,而反应速率的差异是由指前因子造成的,其中反应速率O位 > 邻位 > 对位。邻位甲基化反应的第一代催化剂为氧化铝催化剂4,该催化剂存在选择性低、副反应多等明显缺陷。第二代催化剂为氧化镁催化剂5,活性好,选择性高,但是反应温度较高(600 °C),催化剂易积炭而失活。Velu等6采用共沉淀法制备了镁铝系水滑石催化剂,通过改变镁和铝的比例来调控催化性能。他们发现当Mg/Al为3 : 1时,催化剂表现出最高的活性以及2,3,6-三甲基苯酚选择性,但是2,3,6-三甲基苯酚的选择性也只有 40%。第三代催化剂是以Fe2O3为主要成分的复合氧化物催化剂7。该催化剂有着良好的活性(转化率> 95%)和选择性(>90%),而且操作温度(约 350 °C)较低,寿命长,是目前工业过程中最常用的催化剂。Xiao等8开发了一种以铁氧化物为主的复合氧化物催化剂,该催化剂在间甲苯酚邻位甲基化反应中性能优良,间甲苯酚转化率为 100%,2,3,6-三甲基苯酚的收率达到99.9%,寿命达3000 h以上。该课题组还探究了K2O对该反应的影响9,发现随着K加入量的增加,邻位甲基化的选择性先增加后减少,催化活性起初变化不大,当 K/Fe摩尔比大于0.006时,活性就逐渐降低。出现这种现象主要源于K2O的给电子作用,使得反应底物的吸附变强,然而过多的K2O会覆盖活性位点导致活性的降低。他们还提出了相应的反应机理(图4)。Gandhe等10,11研究了金红石型的TiO2对邻位甲基化反应的性能,发现选择性可达100%。他们认为这主要由于底物吸附构型造成的(图5),苯酚在金红石TiO2表面呈垂直状吸附,表面原位产生的甲基正离子只能与酚的邻位接触,所以导致100%的选择性。而催化剂表面的酸碱对会对苯酚及其衍生物的吸附造成很大的影响12,13。此外,还有关于Mn及Cr的氧化物催化该反应的研究14。总的来说,甲基化反应的活性中心应为酸性位,这在早期的有机反应中已有记载,B酸或者L酸都具有催化活性;碱性位的引入可以改变酚的吸附方式,从而进一步提高选择性;但是引入过量碱性助剂可能会导致活性下降,所以二者之间需要权衡。
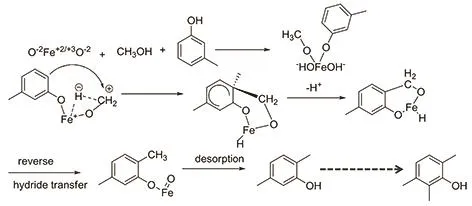
图4 铁氧化物催化间甲苯酚邻位甲基化的反应机理9Fig. 4 The mechanism of o-methylation of m-cresol catalyzed by iron oxides 9.
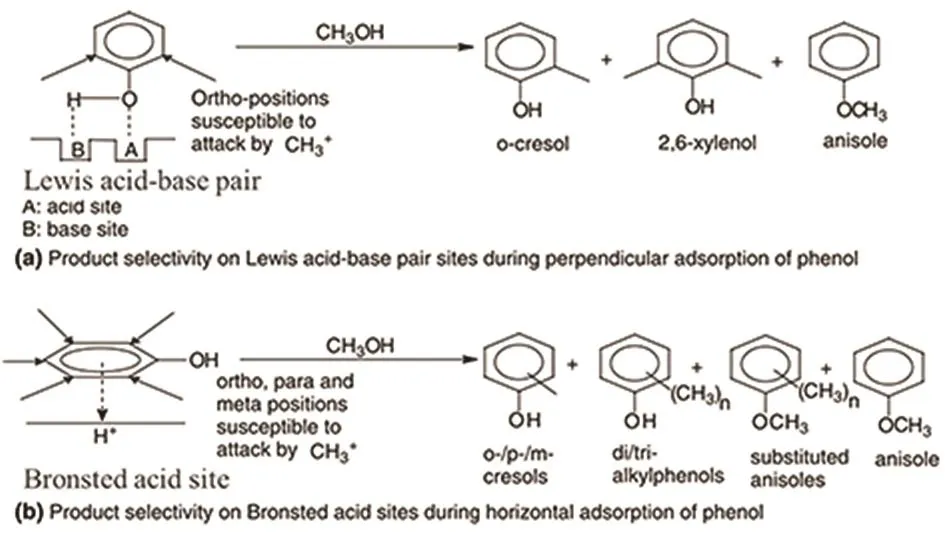
图5 苯酚在不同催化剂表面的吸附方式10Fig. 5 Adsorption models of phenol on defferent sites 10.
由 2,3,6-三甲基苯酚氧化到 TMBQ可分为电氧化法、化学氧化法及催化氧化法。电氧化法由于能耗高,收率低,污染大等因素,不适合工业生产,相关研究也较少。化学氧化法是先将2,3,6-三甲基苯酚磺化,再经氧化剂(常用的为MnO2)氧化得到TMBQ15。该方法污染较大,对设备的腐蚀严重,已经逐渐被淘汰。在该工艺中使用较为温和的氧化剂H2O2并引入催化剂,可有效地解决收率低、污染大等问题。该过程最早使用的是以 CuCl2为主的均相催化体系16。但是想达到较好的催化效果,需要加入一定量的助剂,比如LiCl17、MgCl218等。然而 CuCl2催化剂最大的问题是活性低,往往需要化学计量甚至更多的催化剂来达到很高的催化活性。钱东等计算了CuCl2-LiCl体系下的催化剂用量,得出每氧化1 mol的2,3,6-三甲基苯酚,则需要1–2 mol的 CuCl2和约 4 mol的 LiCl。这个缺陷被日本学者19–21克服,他们发现加入一定量的有机胺或者氯化羟胺,得到同样催化活性需要CuCl2的量可减少至原来的十分之一。Sun等22以离子液体[BMIm]Cl反应介质,CuCl2为催化剂,氧气为氧化剂来实现2,3,6-三甲基苯酚的氧化,共溶剂正丁醇的使用可提升催化活性和选择性,TMBQ的产率高达86%。该过程的最大优点在于只需要催化量的CuCl223,而且活性中心的结构被精确表征出来(图6),他们还提出了如图7所示的反应机理。Li等24,25发现离子液体的阴离子和阳离子都会影响选择性,TMBQ的选择性受阴离子的酸碱性影响很大,其中中性阴离子选择性最高(94.7%),具体表现为选择性 BF4−> Cl−> H2PO4−>HSO4−> OAc−,与阴离子相比,阳离子的影响较弱,侧链所含C原子数目越多,选择性越低。离子液体具有双重作用,既可以与金属形成配合物改变金属中心的电子结构,也可以作为溶剂,增强底物和催化剂的溶解度,加快传质过程。虽然催化剂的用量问题得以有效解决,但是该催化剂毕竟属于均相催化体系,这就不可避免的存在分离困难、催化剂流失等问题。因此,多相催化体系逐渐进入人们的视野。经历早期对杂多酸的均相催化研究26,27之后,Qian等28将磷-钼-钒杂多酸催化剂负载到酸处理后的活性炭和 γ-Al2O3上,得到 TMBQ的收率高达58%。Kholdeeva等26发现在杂多酸均相反应体系中,VO2+是催化反应的活性中心。Palacio等29采用溶胶-凝胶法制备了V2O5-SiO2复合氧化物催化剂,用于2,3,6-三甲基苯酚的液相氧化,转化率最高可达90%,TMBQ的选择性高达99%。活性中心在该过程中的流失可以忽略,催化剂表现出良好的循环使用性能。而且通过加入自由基捕捉剂来证实了这一过程遵循自由基机理。近年来,Ti基催化剂逐渐成为催化氧化领域的热点,其中钛硅分子筛TS-1应用广泛。但是TS由于较小的孔道,使得分子扩散较慢,易滞留导致孔道堵塞而失活。随后出现的钛硅介孔分子筛,有效地解决了这个问题,提高了TMBQ的收率30。但水的存在会破坏某些分子筛的结构,影响催化剂的稳定性31,这严重影响了其工业化前景。Zhou等32采用蒸汽辅助结晶法得到一种多级孔道的介孔钛硅分子筛c-Ti-TUD-1,相比于传统的TS-1该催化剂表现出较好的水热稳定性和催化活性,这主要源自于它本身的多级孔道结构。Kholdeeva等33采用溶胶-凝胶法制备了 TiO2-SiO2气凝胶,可以100%的氧化 2,3,6-三甲基苯酚到 TMBQ,还提出了“双位点”机理(图8),认为介孔结构、Ti物种的高分散性和亲水表面有利于TMBQ的生成,此外作者还对反应条件进行分析,指出以下最优条件:(1) 使用弱配位能力的溶剂(乙腈)有利于 2,3,6-三甲基苯酚在Ti位点上的吸附;(2) 温度为80 °C;(3) 底物浓度不超过 0.1 mol·L−1;(4) 过氧化氢与底物的摩尔比约为3.5;(5) 底物与Ti的摩尔比要小于20。但是材料本身较差的稳定性限制了其应用。除了传统的多相催化外,最近光催化在氧化2,3,6-三甲基苯酚到TMBQ方面也有应用,所使用的催化剂主要有CuO34和尖晶石类材料35,36。该过程虽然有着高选择性(≈ 100%),但其较低的转化率是急需解决的难题。目前2,3,6-三甲基苯酚氧化到TMBQ的工艺仍然存在很多问题,多数催化过程需要使用H2O2这种氧化剂,如何使用氧气甚至空气来实现该过程的高效转化会是今后发展的重点之一37。
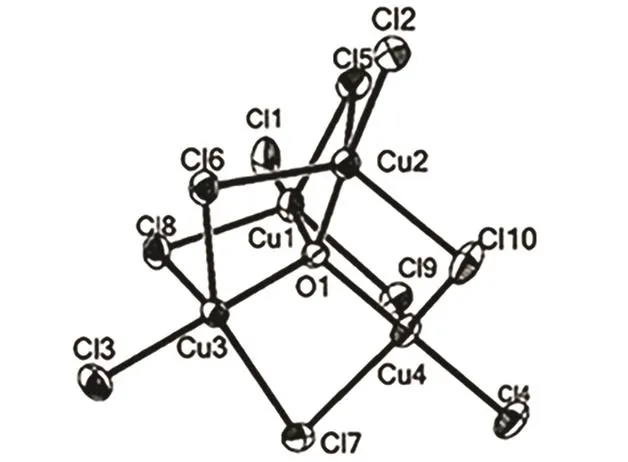
图 6 [Cu4(µ4-O)Cl10]−4的结构 22Fig.6 The structure of [Cu4(µ4-O)Cl10]−4 22.
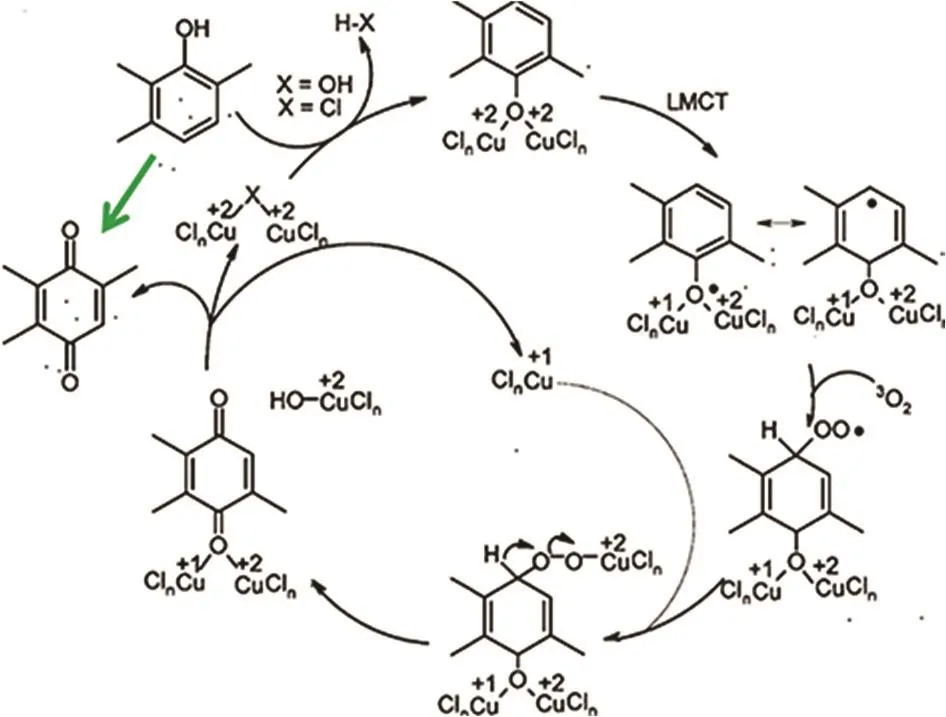
图7 2,3,6-三甲基苯酚氧化至TMBQ的反应机理22Fig. 7 Proposed mechanism of 2,3,6-trimethylphenol oxidation to TMBQ 22.
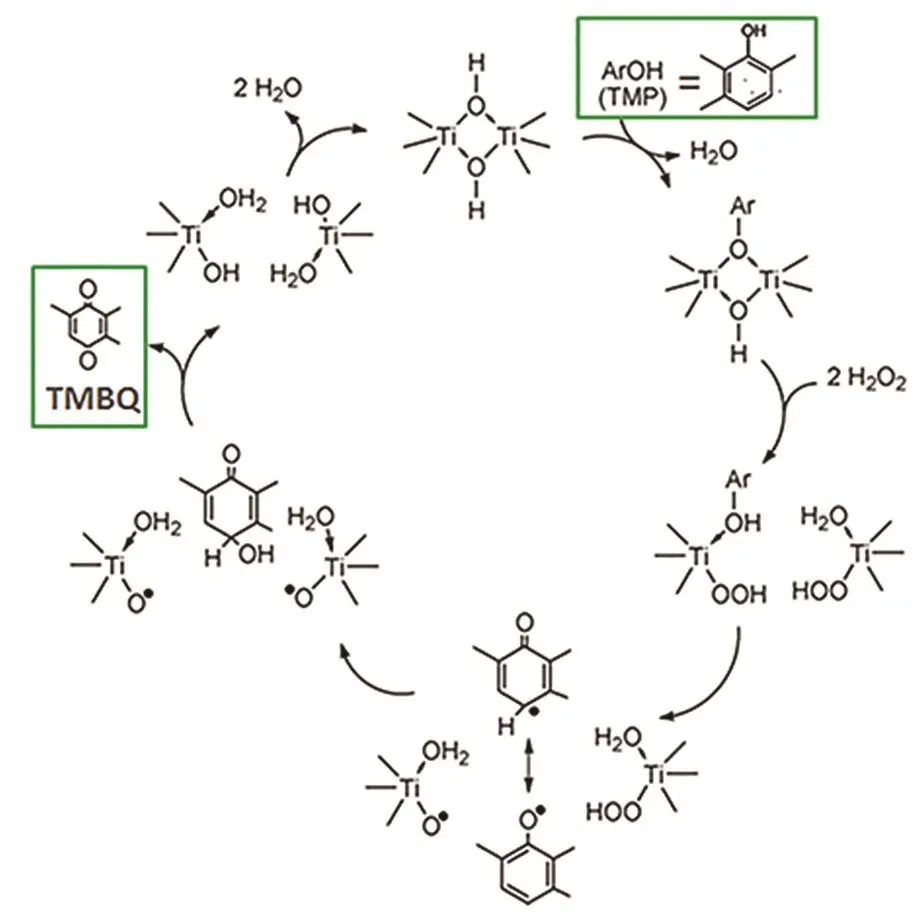
图8 Ti基催化剂上2,3,6-三甲基苯酚氧化为TMBQ的“双位点”机理33Fig.8 Hypothesized mechanism of 2,3,6-trimethylphenol oxidation to TMBQ over a ‘double site’ 33.
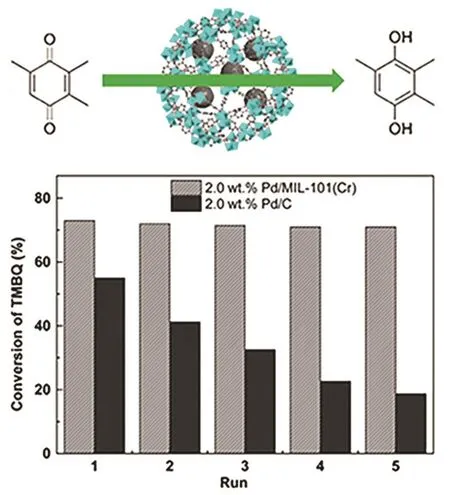
图9 Pd催化剂在TMBQ催化加氢反应中的套用性能42Fig. 9 Reusability of Pd-based catalysts in the catalytic hydrogenation of TMBQ 42.
三甲基苯醌还原到三甲基氢醌传统的方法是用保险粉为还原剂38,该工艺需要化学计量甚至更多的还原剂,导致污染大、成本高。目前工业上常用以H2为还原剂,Pd/C为催化剂的催化加氢工艺。Zhang等39报道了一种以LBA(商业化的混合溶剂)为溶剂,以Pd/C为催化剂的加氢工艺,90 °C、0.3 MPa H2的条件下,TMHQ 的收率为99.4%,产品纯度为96.7%。催化剂较为稳定,失活的原因归结为底物和产物在表面的强吸附。在以Al2O3为载体时,催化剂的焙烧温度对活性影响很大,焙烧温度过高会导致活性的下降,最好控制在 300–500 °C40。Qian 等41通过正交实验确定了Pd/Al2O3催化剂的最佳实验条件为50 °C,H2压力0.1 MPa,TMBQ空速为 0.27 h−1,产品无需后处理,TMHQ的纯度可达98%以上。Zhao等42将Pd纳米颗粒负载于MIL-101(Cr)的孔道内,粒径约为2 nm,相比于Pd/C催化剂,套用性能明显提升(图9),主要由于MIL-101(Cr)对Pd纳米颗粒有很好的限域作用,使得其难以团聚或者流失。Raney Ni作为一种古老的加氢催化剂,也常被应用于 TMBQ的加氢。Zhang等43通过正交实验确定了较优的条件:底物初始浓度为0.1 g·mL−1,催化剂用量为底物的 10%,H2压力为 0.7–0.8 MPa,温度为 100 °C(转速 8000 r·min−1)。这样的条件下,TMHQ 的收率为 97.3%,产品纯度为 93.1%。Mukhopadhyay等44研究了Raney Ni催化TMBQ加氢的动力学过程。结果发现该过程对Raney Ni是一级过程,Langmuir-Hinshelwood模型可以很好地解释动力学现象,计算得到的活化能为 52 kJ·mol−1。最近本课题组45用一步煅烧法制备CoOx@CN催化剂,在100 °C、2 MPa H2条件下,TMBQ的转化率高达90%,TMHQ选择性99%以上。通过DFT计算表明金属态的Co是加氢的活性位点,高选择性的原因在于产物TMHQ的脱附能垒(0.33 eV)比进一步加氢能垒(0.57 eV)低(图10)。该工作引领了廉价纳米金属在TMBQ选择性加氢方面的应用。廉价金属的使用可以降低催化剂成本,为工业化提供一种选择。
2.2 对叔丁基苯酚法
最近能特科技股份有限公司开发了一种从对叔丁基苯酚到 2,3,6-三甲基苯酚的路线46,2,3,6-三甲基苯酚可按照前述氧化-还原等方法(详见2.1)得到TMHQ,如图11所示。
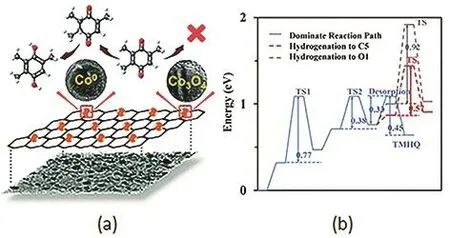
图10 (a) CoO x@CN催化TMBQ加氢的示意图;(b)TMBQ加氢过程中反应物、中间态及产物在Co(111)表面的势能变化图45Fig. 10 (a) The diagram of TMBQ hydrogenation catalyzed by CoO x@CN; (b) Potential energy profiles of TMBQ hydrogenation on the Co(111) 45.
该工艺的第一步是将对叔丁基苯酚与甲醇通入反应器,以铁氧化物为催化剂,得到 2,6-二甲基-4-叔丁基苯酚;第二步将 2,6-二甲基-4-叔丁基苯酚与甲醇通入反应器,以氧化铝为催化剂,得到2,3,6-三甲基-4-叔丁基苯酚;然后用 1%的硫酸将叔丁基脱掉,精馏得到2,3,6-三甲基苯酚。这个工艺采用较为易得的对叔丁基苯酚,有着很好的经济效益,尤其在目前间甲苯酚原料紧缺的情况下,有着很好的发展前景。
2.3 苯酚法
由苯酚到 TMHQ需要三步(图 12):(1) 与甲醇发生甲基化反应产生2,4,6-三甲基苯酚(TMP);(2) TMP氧化生成4-羟基-2,4,6-三甲基-2,5-环己二烯酮(TMCH,或者叫甲代二氢醌);(3) TMCH发生甲基转位反应生成 TMHQ。该方法的一个特点是TMP既可由苯酚甲基化得到,也可直接从生产2,6-二甲基苯酚(合成 PPO 耐热树脂的单体)工艺中的副产物分离提纯得到47。这样一个过程可以有效地利用工业副产品。
苯酚甲基化生成TMP的反应与前述邻位甲基化(2.1.1节)相似之处在于都是酸催化的甲基化反应。区别是这里需要实现酚对位的甲基化,反应相对简单易行。MgO是常用的催化剂48,还可以添加 Ga2O3组分49。反应温度为 400–500 °C。第二步TMP到TMCH的氧化反应通常需要在碱性介质中反应,可以是水或者有机溶剂。常用的催化剂包括氧气或者空气50,51、次卤酸及其盐类52–54和二氧化锰55,56(需加质子酸助剂)。不加碱的情况下,氯气也可以直接完成这个反应,不过会产生大量的氯化氢57。该反应条件较为温和。第三部转位反应的条件非常简单,直接加热(≥ 100 °C)即可58。
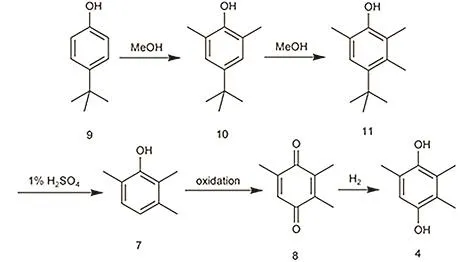
图11 对叔丁基苯酚法制备TMHQ的工艺路线图Fig. 11 The preparation of TMHQ by using 4-tertbutylphenol as a source.
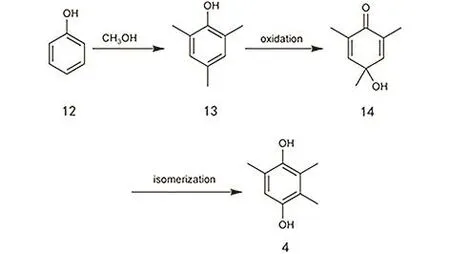
图12 苯酚法制备TMHQ的工艺路线图Fig. 12 The preparation of TMHQ by using phenol as a source.
能特科技股份有限公司报道的工艺同样以苯酚为原料,先后经历邻位甲基化、磺化、酯化、间位甲基化、脱磺酸基等反应,最终得到2,3,6-三甲基苯酚产品59。
2.4 二乙基酮法
二乙基酮法的反应路径如图13所示,与二乙基酮反应的试剂可以是甲基乙烯基酮,也可以是巴豆醛,它们都会先历经羟醛缩合,随后环化,最后脱氢形成 2,3,6-三甲基苯酚。该反应需要碱催化,在反应体系中会加入一定量的碱试剂,比如苄基三甲基胺氢氧化物,反应温度为 50–250 °C之间。在实际生产中,多数情况所加入的反应物并非甲基乙烯基酮或者巴豆醛,而是可以原位转化为它们的试剂60,61。1-胺基-2-乙烯基甲基酮(图14)是最常用的反应试剂62,加入量(摩尔量)通常是二乙基酮的0.1–1倍,需要强调的是,在这样的条件下胺基可以作为一个离去基团,那么在环化步骤中双键可以保留,因此无需脱氢即可得到2,3,6-三甲基苯酚,1-胺基-2-乙烯基甲基酮的转化率可达89%。
2,3,6三甲基苯酚可经过氧化、加氢等步骤得到 TMHQ(详见 2.1)。
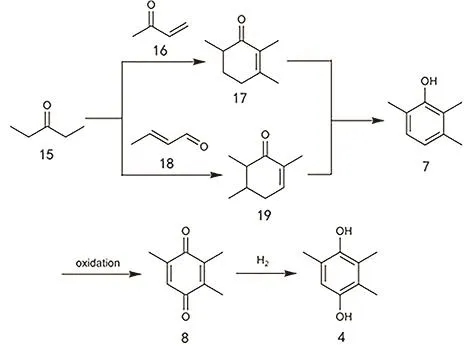
图13 二乙基酮法制备TMHQ的工艺路线图Fig. 13 The preparation of TMHQ by using diethyl ketone as a source.
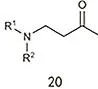
图14 1-胺基-2-乙烯基甲基酮的分子结构Fig. 14 The structure of 1-amino-2-vinyl methyl ketone.
2.5 偏三甲苯法
如图15所示,该方法以偏三甲苯为原料,先后经历磺化、硝化、硝基的还原、水解脱磺酸跟、氧化可得TMBQ63,然后TMBQ在Pd/C催化下加氢得到TMHQ。作为合成TMHQ的传统工艺,它采用的偏三甲苯来源于催化重整副产物C9重芳烃,因此廉价易得,成本低。但它存在明显的缺陷即工序多,产率低且污染严重,目前已经基本淘汰。
近年来,越来越多的研究集中于偏三甲苯直接氧化制备 TMBQ或者 TMHQ。Bao等64采用Fenton试剂来氧化偏三甲苯到TMBQ,发现偏三甲苯与H2O2摩尔比1 : 1的状况下,二价铁的投料量为 4倍时,反应效率最佳,45 °C下 5 h内TMBQ的收率为34.95%。H2O2-CH3COOH-H2SO4是较为常用的一种反应体系,Chen等65通过正交试验确定了该反应体系的最佳条件:H2O2与偏三甲苯的摩尔比为6.5 : 1,添加H2SO4的摩尔量是偏三甲苯的3倍,反应温度70 °C,在该条件下反应3 h,产物TMBQ的纯度可达92.13%。Zhao等66采用同样的反应体系,加入Cu-MCM-41为催化剂,偏三甲苯的转化率78.86%的情况下,TMBQ的选择性可达83.86%,剩下的则为TMHQ。Zhang等67,68对γ-Al2O3进行修饰改性,并探究其催化氧化偏三甲苯到 TMBQ的催化性能,由于 γ-Al2O3有着廉价易得等优势,因此有着较好的应用前景。
目前无论采用哪一种催化剂,直接氧化法都必须采用H2O2这种较为昂贵的工业氧化剂,因此廉价氧化剂(比如空气)的应用是取得突破的关键之一。
2.6 5-异丙基偏三甲苯法
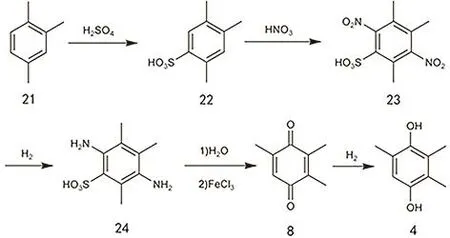
图15 偏三甲苯法制备TMHQ的工艺路线图Fig. 15 The preparation of TMHQ by using 1,2,4-trimethylbenzene as a source.

图16 5-异丙基偏三甲苯法制备TMHQ的工艺路线图Fig. 16 The preparation of TMHQ by using 5-isopropyl-1,2,4-trimethylbenzene as a source.
以5-异丙基偏三甲苯为原料,需经过磺化、碱熔和脱异丙基才能得到TMHQ (图16)。该工艺相对简单易行,存在的主要问题是原料不纯,目前5-异丙基偏三甲苯原料中含有17%的6-异丙基偏三甲苯不易分离,因此反应会有大量副产物,这严重限制了该方法的应用。
2.7 异氟尔酮法
丙酮首先聚合为α-异佛尔酮,之后重排为β-异佛尔酮,然后氧化为茶香酮(KIP,氧代异佛尔酮),也可直接由α-异佛尔酮氧化到KIP,KIP随后重排酰化、皂化水解得到TMHQ (图17)。或者在重排酯化得到二乙酸三甲基氢醌(DMHQ-DA)后,直接与异植物醇反应可得到乙酰生育酚。该方法是近年来出现的新工艺,原料廉价易得、工艺简单、污染小,是一种高效环保的生产工艺。
异佛尔酮存在 α和 β两种异构体,α异构体较为稳定,由α到β的转变并不难,所用到的催化剂有金属69、金属氧化物及一些混合氧化物70,71。
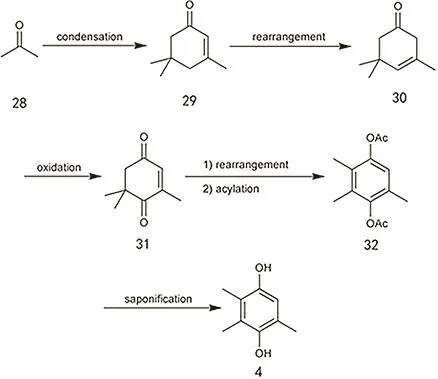
图17 异氟尔酮法制备TMHQ的工艺路线图Fig. 17 The preparation of TMHQ by using isophorone,produced by trimerization of acetone, as a source.
β-异佛尔酮的氧化是该工艺的关键技术之一。目前研究较多的催化剂有过渡金属的无机盐及配合物、金属氧化物和一些无金属体系。大多数反应体系需要添加碱性助剂72,73,这些试剂的加入会造成活性中心的流失等问题74,不过使用叔胺可以很好地解决这个问题。在众多催化体系中过渡金属乙酰丙酮盐是研究较早的一类75。Li等76发现咪唑盐类的加入可以提升乙酰丙酮铁的活性和稳定性。Chen等77分析了该反应的动力学过程,发现它对 β-异佛尔酮是准一级反应,生成产物 KIP的活化能约为 70.5 kJ·mol−1,指前因子约为 33.53,而生成主要副产物4-羟基-3,5,5-三甲基-2-环己烯-1-酮(HIP)的活化能约为 86.4 kJ·mol−1,指前因子约为36.23。Sorokin等78研究了酞菁铁催化体系对于 β-异佛尔酮氧化的性能,但是该反应体系需三乙胺的参与。随后该课题组79以壳聚糖形成的微球为载体,并将酞菁铁(或者钴)负载其上,用于β-异佛尔酮的氧化,由于壳聚糖上氨基表现出的碱性,使得该反应体系不需要添加碱性助剂。Burns等80发现在一系列金属的四苯基卟啉配合物中,Mn的对应催化剂活性最高,而且反应中异构产生α-异佛尔酮的量很少,原因在于 Mn中心与 β-异佛尔酮独特的结合方式,并且他们提出了两种反应机理(图 18)。Thatte等81以金属(Mn、Cu、Co)席夫碱配合物为催化剂,以空气为氧化剂,可高效实现 β-异佛尔酮的选择性氧化,转化率高达94.1%,KIP的选择性为83%。Li等82研究了Cu、Ce和Nd三种金属的席夫碱化合物催化该反应的选择性大小顺序,结果为 Cu(II) > Ce(IV) > Nd(III),其中 Cu(II)席夫碱化合物得到的 KIP选择性为90%。本课题组83对C3N4进行修饰,提高其在可见光范围内的吸收,并考察它们光催化 β-异佛尔酮氧化的性能,发现KIP的选择性高达92%,而且NHPI (N-羟基邻苯二甲酰亚胺)的添加可以大大提高活性。
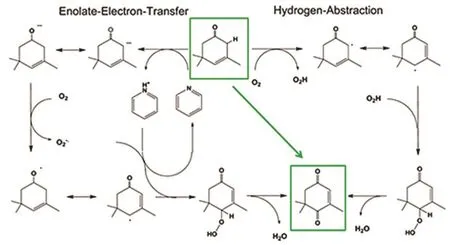
图18 β-异氟尔酮到KIP的两种自由基机理80Fig. 18 Two alternative mechanisms for the radical pathway from β-IP to KIP 80.
α-异佛尔酮热力学上比 β-异佛尔酮稳定,直接氧化到KIP较为困难。Kishore等84,85对镁铝水滑石负载的金属催化剂催化氧化 α-异佛尔酮的性能进行了大量研究,在使用叔丁基过氧化氢为氧化剂的情况下,KIP的选择性可达到100%。由于α-异佛尔酮的稳定性,它的氧化往往较为苛刻的反应条件,Li等86,87采用 NHPI为催化剂,可以在较为温和的条件下实现α-异佛尔酮到KIP的转化。Zhong等88报道了一个均相催化剂体系,CuCl2为催化前驱体,乙酰丙酮为溶剂,他们认为原位形成的Cu(acac)Cl2是起催化作用的关键物质,而且以烯醇式配位形成的物种活性更高,但是反应产生的水会导致催化剂失活。该反应鲜有气相催化的报道,Murphy等89研究了钒氧化物催化气相氧化α-异佛尔酮的性能,发现KIP的选择性较低,主要的副反应是环外侧链甲基的氧化,而且催化剂的活性随着反应时间一直降低,原因是积炭以及V价态的降低。虽然300 °C以上可以解决失活的问题,但是又会造成底物的深度氧化。
KIP的重排酰化是该工艺中另一个关键的步骤,传统的方法采用B酸或者L酸为催化剂90–92,这类催化剂活性高,但存在腐蚀性强,产品分类困难等问题。随后固体酸的发展受到关注,有ZnCl293、InCl394、杂多酸95以及全氟磺酸树脂96。Rac等97将丙磺酸和苯磺酸嫁接到介孔二氧化硅上,这些催化剂对于KIP的重排酰化活性很低,而且只会得到单酰化的产物。Nafion的催化性能稳定,但活性较低。Hinze等98将Nafion与二氧化硅结合,大大提升了其热稳定性,为工业化应用打下了基础。
3 异植物醇的合成
异植物醇是构成维生素E的另一个结构基元,它是由四个异戊二烯分子首尾相接而构成的双萜类的二十个 C不饱和烯叔醇,其结构如图 19所示。它其实包含三个手性中心,但是手性催化并不是本文讨论的范围。本文将讨论如何构筑这个含有20个C的分子。纯净的异植物醇为无色油状液体,有很弱的花香和香脂香气,不溶于水,易溶于醇、酮等有机溶剂。
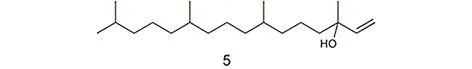
图19 异植物醇的分子结构Fig. 19 The structure of isophytol.
根据制备过程中重要中间体的不同,异植物醇的主要合成工艺可分为假紫罗兰酮工艺和芳樟醇工艺。
3.1 假紫罗兰酮工艺
假紫罗兰酮工艺可采用山苍子油(主要成分为柠檬醛)为原料,是制备异植物醇较为经典的方法,早在1967年就有相关专利99。如图20所示,该工艺首先将柠檬醛与丙酮发生 Claisen-Schmitt反应,生成假紫罗兰酮,这是一个由碱催化的反应,可以用碱液,也可用固体碱。最近镁铝水滑石类材料受到广泛研究。水化是一种提高其活性的主要方法100。高温煅烧后的材料表面碱性位更稳定,但活性要比水化后的材料低。这类材料表面的碱性位在空气中很快会被 CO2覆盖,而且反应底物的强吸附也会导致催化剂失活,这也是其工业化应用的主要障碍101。Dıez等102发现反应速率随催化剂表面O2−强碱性位的多少线性增加,而且丙酮的自缩合是主要的副反应之一,它的速率随着 L酸位点增加而增加。在镁铝水滑石表面负载碱金属可以提高其催化活性103。Di Cosimo等104研究发现,Na、K、Cs三种碱金属由于离子半径太大,导致MgO孔道被堵塞,活性下降。而半径较小的Li则不同,当Li的负载量低于0.5% (w,质量分数)时,MgO的碱性随着Li负载量的增加而增加,从而是活性上升;当 Li的负载量高于0.5% (w)时,Li物种的团聚以及Li2CO3的形成都会导致活性的下降。稀土元素在该反应中也有应用105,Wang等106发现镁铝混合氧化物在Y表面修饰后活性大大提高,而La和Eu都没有这种效果,原因是Y改变了母体氧化物的孔道结构和比表面积,使得更多的碱性位暴露。Raju等107将KF/γ-Al2O3用于该反应中,得到假紫罗兰酮的选择性为 93%,并且他们将活性与 KF的(111)面关联起来。但由于受到天然原料山仓子油资源的限制,生产规模较小,不能满足市场需求,近年来该方法已逐渐被淘汰。由假紫罗兰酮到异植物醇的具体方法与芳樟醇工艺相同,会在芳樟醇工艺中详细介绍。
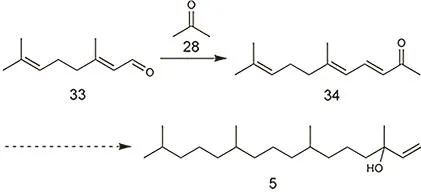
图20 假紫罗兰铜工艺制备异植物醇Fig. 20 Preparation of isophytol via pseudoionone.
3.2 芳樟醇工艺
芳樟醇工艺是目前世界上绝大多数异植物醇生产装置采用的工艺,通常需要从甲基庚烯酮(6-甲基-5-庚烯-2-酮)而来。目前生产甲基庚烯酮主要有两条路线:乙炔-丙酮法(Roche法)和丙酮-异丁烯-甲醛法(BASF法),如图21所示。乙炔-丙酮法以乙炔和丙酮为初始原料,先得到甲基丁烯醇(2-甲基-3-丁烯-2-醇),再通过反应得到甲基庚烯酮。而丙酮-异丁烯-甲醛法108将丙酮、异丁烯和甲醛一步反应得到6-甲基-6-庚烯-2-酮,它可转位为甲基庚烯酮,也可以氢化得到6-甲基-2-庚酮进行后续的反应。从甲基庚烯酮出发得到异植物醇的过程是一个碳链增长的过程,虽然步骤较多,但只涉及两个反应,以甲基庚烯酮到香叶基丙酮为例,先是甲基庚烯酮产生芳樟醇(反应A),随后芳樟醇转化为香叶基丙酮(反应 B)。其实乙炔-丙酮法生产甲基庚烯酮也是先后经历A、B两个反应。
反应 A通常有格式法和催化加氢法两种。格式法是将甲基庚烯酮与含乙烯基的格式试剂(H2C=CHMgX)反应,然后质子化即可得到芳樟醇99。但该方法涉及有机试剂的使用,操作复杂,成本高,已经逐渐被淘汰。催化加氢法是先将甲基庚烯酮与乙炔反应得到去氢芳樟醇,然后去氢芳樟醇选择性加氢得到芳樟醇。整体上可以归纳为甲基酮的乙炔化反应和炔醇半加氢反应两个部分。
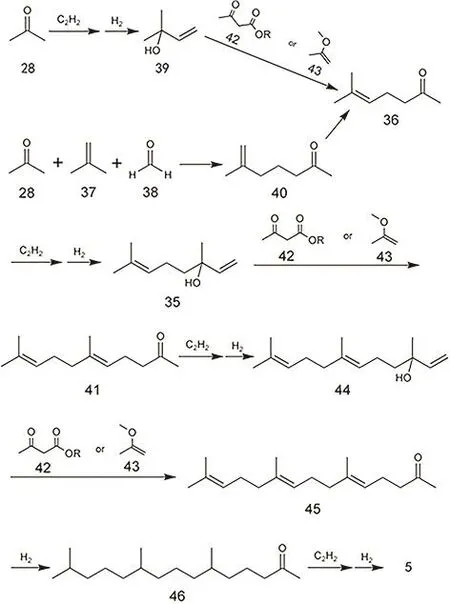
图21 异植物醇的两种主流合成工艺Fig. 21 Two main synthetic processes for preparing isophytol.
甲基酮的乙炔化反应是一个由碱催化的亲核加成反应,是一个很简单的有机反应,该体系常用的催化剂为NaOR、KOR等碱性试剂109。其实该过程中参与反应的主要是乙炔钠(以采用NaOR为催化剂的体系为例),即乙炔负离子进攻显电正性的羰基C原子,随后羰基氧质子化可得到炔醇的结构。
炔醇半加氢是近年来研究最活跃的领域之一,也是整个异植物醇合成工艺中最关键的一个反应。广义上说,这是一个C≡C选择性加氢到C=C双键的过程,这在聚合物生产工艺里是一个非常重要的过程,因此被广泛研究。在异植物醇的合成工艺中,底物的官能团并不复杂,只存在羟基这种影响很小的官能团,所以很多经典的炔烃选择性加氢催化剂都可以直接应用到这里。Pd催化剂在这一领域受到广泛关注,但通常只能在较低转化率条件下得到较高的选择性110。炔醇半加氢是一个对催化剂结构很敏感的反应。Kiwi-minsker等111发现炔醇半加氢主要发生于Pd的平面位,而过加氢则主要发生于边角位,所以在立方体、八面体、十四面体三种结构的Pd颗粒中,立方体结构由于含有较少的边角位从而表现出最高的半加氢选择性,而且动力学拟合指出3–5 nm的立方体Pd颗粒将会是最优的半加氢催化剂。随后该课题组112又合成了不同粒径(2.1–9.8 nm)的Pd纳米颗粒,分析在炔醇半加氢反应中的尺寸效应,发现随着颗粒变大,炔醇转化率逐渐增加,选择性几乎没有变化(图 22)。他们又以 Langmuir-Hinshelwood模型来分析动力学过程,认为出现这种现象(颗粒越大,活性越高)的原因在于底物的吸附状态,颗粒越小,底物炔醇的吸附越强,导致反应活性较低,这也就是常说的“底物抑制”现象。如果颗粒继续增大,活性的变化将变缓,选择性的改变依然很小113。传统的Lindlar催化剂114采用Pb为助剂,Tschan等发现115以Pd81Si19为催化剂时,Pb对于选择性的调控不如有机物,在有机物中含 S化合物的作用最为明显。但是这个催化体系的高选择性也仅仅出现在炔醇底物低转化率的情况下116,这也是很多催化体系的常见问题,即转化率较高的时候过加氢难以控制,这对于实际生产并不是一个很好的选择。工业上乙炔半加氢使用的是PdAg催化剂117,Kiwi-Minsker等118发现Ag的加入可使得Pd催化炔醇半加氢的选择性由78%增加到93%。此外,Zn或者ZnO是另一类研究较多的助剂,早在1972年就有专利介绍可溶性Zn化学物可以提高 Pd催化炔醇加氢的选择性直至 100%119。Okhlopkova等120采用溶胶-凝胶法,以F127为模板,合成了PdZn/TiO2催化剂,该催化剂在炔醇转化率 95%的情况下,烯醇选择性可达 81.5%–88.9%,要明显高于传统浸渍法得到的催化剂。最近本课题组先是发现 Pd/mpg-C3N4对于苯乙炔加氢到苯乙烯有着很好的选择性121,随后又合成了PdZn/CN@ZnO催化剂,发现它在炔醇选择性加氢方面有很好的性能(图23)122。CN和ZnO存在一种协同作用可以提升反应活性,在高温还原条件下,由于Zn物种的迁移会形成PdZn合金相,理论计算说明Zn可以堵住Pd颗粒的边角位,影响底物、产物及中间态的吸附,从而增加了选择性。Bonrath等123–125开发了一种核壳式的载体,内核是含大量 Fe的合金结构,外面包裹一层氧化物(ZnO或者Al2O3及其混合物),并将Pd或者PdM(M是另一种金属)负载其上用于炔醇的半加氢。Grjaznov等126设计了一种含有PdRu合金的膜催化剂,目标产物烯醇的产率可达96%–99%,优势在于无需催化剂的分离和回收。
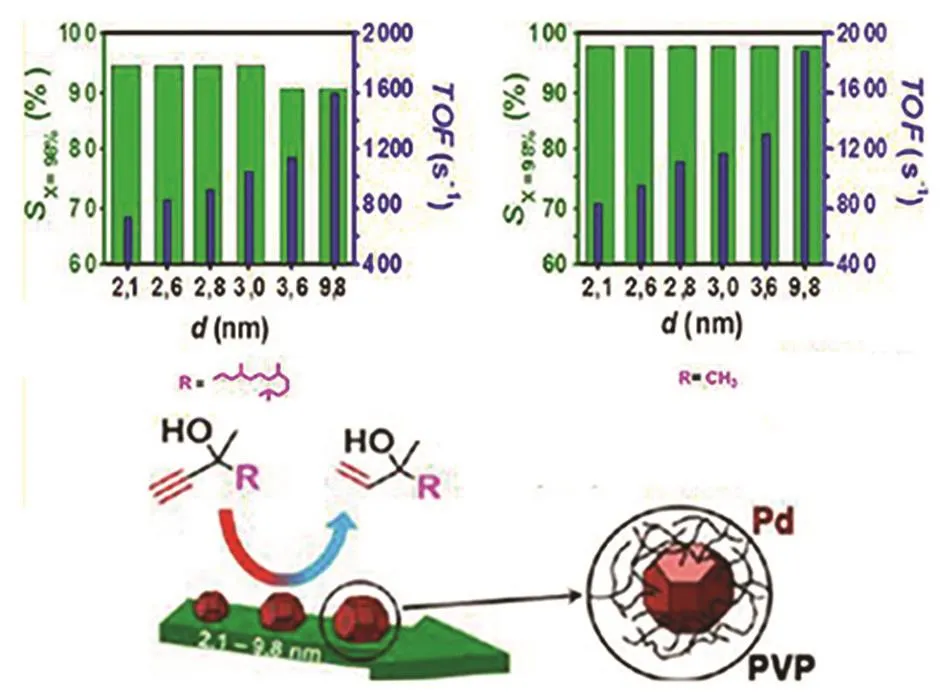
图22 Pd纳米晶催化炔醇加氢的尺寸效应112Fig. 22 Size effect in hydrogenation of alkynol catalyzed by Pd nanocrystals 112.
反应B目前有乙酰乙酸酯法和甲氧基丙烯法两种127。以芳樟醇(也称哪醇)为例,乙酰乙酸酯法(图24)是将芳樟醇与乙酰乙酸酯(通常是乙酰乙酸甲酯,双烯酮也可作为酯化试剂)发生酯交换反应,产生乙酰乙酸芳樟酯,随后该酯发生 Carroll重排128生成香叶基丙酮。Carroll重排通常采用Ru的配合物为催化剂,尤其是涉及到手性合成的时候129–131。Bizet等132提出了由碱催化的 SN2-脱羧的串联反应机理。De Castro等133研究了一系列Lewis酸催化Carroll重排的性能,发现异丙醇铝是最优的催化剂。Oost等134指出异丙醇铝催化该反应的选择性高达97.4%,产率有93%。而甲氧基丙烯法(图 24,也被称为Saucy-Marbet反应)是将芳樟醇与甲氧基丙烯反应生成芳樟醇的甲氧基丙烯醚,随后该醚发生Claisen重排生成香叶基丙酮。Claisen重排反应只需简单地加热到200 °C左右即可发生反应,周环[3,3]-σ迁移重排是被广为接受的一种机理。这里涉及的反应都是研究多年的有机反应,简单易行。Saucy-Marbet反应也可以直接以炔醇为原料进行反应135,只是产物中多出一个双键,对后续步骤影响不大,却无需炔醇半加氢反应,提高了反应效率。
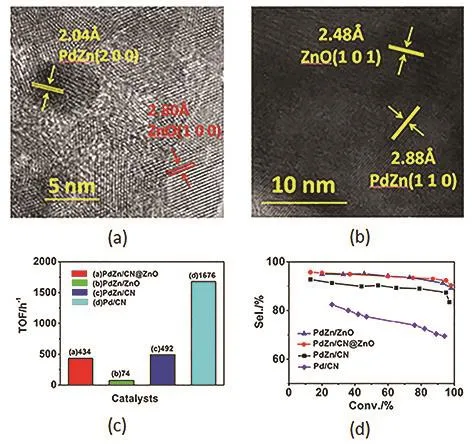
图23 (a,b)Pd Zn/CN@ZnO的HRTEM图像;(c)几种Pd催化剂的炔醇加氢催化活性;(d)几种Pd催化剂催化炔醇加氢的选择性122Fig. 23 (a, b) HRTEM images of Pd Zn/CN@ZnO;(c) TOF values of alkynol hydrogenation by Pd-based catalysts; (d) Selectivity of alkynol hydrogenation by Pd-based catalysts 122.
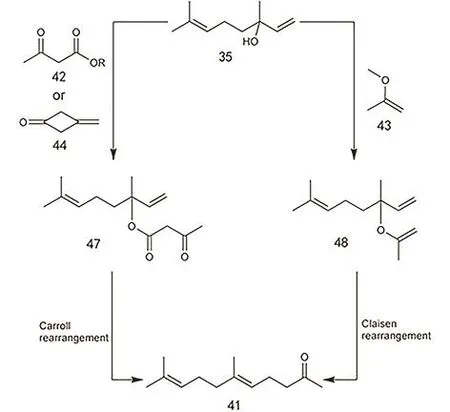
图24 烯醇丙酮化反应的两条路径Fig. 24 Two alternative pathway from linalool to geranylacetone.
3.3 其它工艺
除了上述主流工艺方法外,采用 C5原料构筑异植物醇也是研究较多的方法之一,包括异戊二烯、异戊醛、1-氯-3甲基-2丁烯等。异戊二烯可二聚也可三聚,形成的物质与二甲基亚胺反应后水解可得甲基酮这一构筑异植物醇的中间体(图25)136,137。异戊醛则可以直接和丙酮发生羟醛缩合生成6-甲基-3-庚烯-2-酮这一中间体138。1-氯-3甲基-2丁烯可与4-甲基-3-戊烯-2-酮发生反应,随后产物与乙炔发生乙炔化反应,再进行半加氢,最后经过重排可得甲基酮类物质(图26)139。但该类反应会产生大量含卤废水处理困难,对设备腐蚀严重,因此难以投入实际生产。以上反应都可以在保留局部反应基团的基础上进行一定程度的底物拓展,使用碳链更长的底物进行反应140–142。醛类物质和卤代物都可以由α-烯烃转化而来143。醛类物质反应后得到的酮都会含有C=C,这就需要后续的加氢步骤144,Kramer等145采用醛和丙酮反应,使用负载型Pd催化剂,载体为MgO和Al2O3的混合物,在一定氢气压力下,可以一步得到饱和的甲基酮。加入催化剂可以大大提高目标产物的选择性,否则会存在很多原料的自缩合产物。
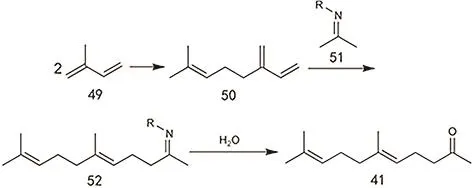
图25 异戊二烯法制备香叶基丙酮的合成路线Fig. 25 One method from isoprene to geranylacetone.
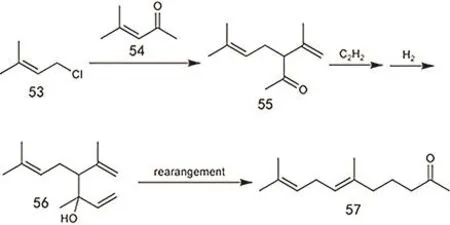
图26 以1-氯-3甲基-2丁烯为原料制备6,10-二甲基-6,9-十一碳二烯-2-酮的合成路线Fig. 26 One method from 1-chlorine-3-methyl-2-butene to 6,10-dimethyl-6,9-undecadiene-2-one.
Teles等146开发了如图27所示的合成路线,以 3,7-二甲基-2-辛烯-1-醇为原料,先后经历酯交换、重排、Claisen-Schmitt反应、选择性加氢等4个步骤可以得到植酮。这样要比同样以C10芳樟醇为原料的工艺步骤简单。
松节油法可利用松节油为原料,先转化为芳樟醇,再转化到异植物醇。松节油是松属植物分泌的松脂经过蒸馏得到的挥发油,约含有60%的α-蒎烯和 30%的 β-蒎烯。早在 1976年,美国 SCM Glidden率先开发了由 α-蒎烯制备芳樟醇的工艺路线(图28),并于1982年正式投产147。芳樟醇可通过前述方法得到香叶基丙酮。其实在松节油的成分中也含有少量的月桂烯,β-蒎烯直接热裂解也可得到月桂烯148,如图29所示月桂烯可经由香叶基氯得到香叶基丙酮,或者经由橙花基氯得到橙花基丙酮149,150,也可直接与丙酮亚胺反应后水解得到香叶基丙酮或橙花基丙酮137。
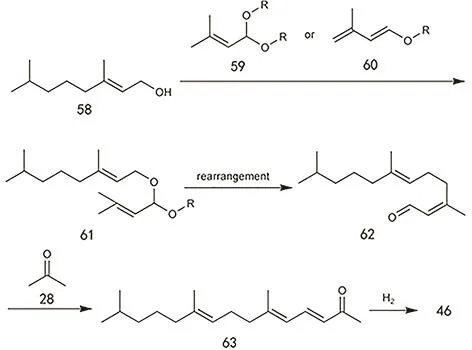
图27 以3,7-二甲基-2-辛烯-1-醇为原料制备植酮的合成路线Fig. 27 One method from 3,7-dimethyl-2-octene-1-ol to phytone.
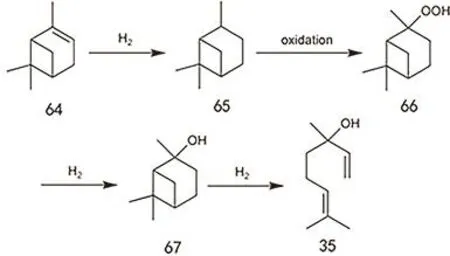
图28 α-蒎烯制备芳樟醇的工艺路线Fig. 28 One method from α-pinene to linalool.
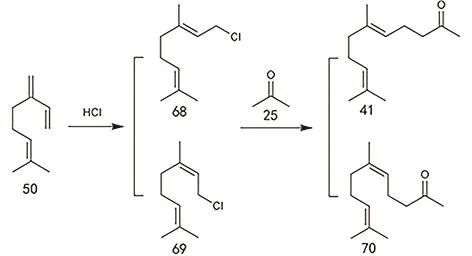
图29 以月桂烯为原料制备香叶基丙酮或橙花基丙酮的合成路线Fig. 29 One method from myrcene to geranylacetone or nerylacetone.
美国Amyris公司最近开发了一种生物法,采用改造酵母可以将糖类以及乙醇转化为多种类异戊二烯类物质,其中包括法尼烯这种生产异植物醇的中间体151–154。
4 生育酚的制备
以TMHQ和异植物醇为原料合成生育酚本质上是Friedel-Crafts烷基化和脱水缩合反应的串联过程,这两个反应都是研究多年的有机反应,是酸催化的反应,B酸和L酸皆可,室温至200 °C均可。如图30所示,呋喃环的形成和异植物醇的直接脱水是两个主要的副反应。而且产生的水会毒化某些L酸催化剂,为了尽量避免副反应发生提高生育酚的产率,可以采用将异植物醇的溶液滴加到TMHQ溶液中的方法。该反应常用的催化体系有金属卤化物、含氟强酸两类物质,但它们都存在分离困难,污染严重等缺点,近年来阳离子交换树脂在该领域内逐渐受到关注155。
ZnCl2/HCl是一类传统催化体系156,157,它面临的主要问题是回收困难、对设备腐蚀严重、污染大等。固载在一定程度上可以缓解这些难题158。最近Kemnitz等159,160采用溶胶-凝胶法将甲醇镁和异丙醇铝与HF的水溶液混合加热得到表面羟基化的MgF2和AlF3,表面羟基的数量直接决定了这些材料的酸性种类和强度。在MgF2的体系中,羟基竟然可以表现出B酸性,而非通常MgO上的碱性,可能是由于F与羟基的氢键作用。这种B酸性质的羟基可以有效地催化TMHQ和异植物醇的缩合。但是羟基过多就会导致碱性的出现,使得催化活性下降。异丙醇铝在50%的HF水溶液中反应得到的AlF3-50表现出最好的催化性能,生育酚的产率达到99.9%以上。纯相MgF2和AlF3都没有催化活性,这表明反应的起始步骤并不是由L酸催化。该课题组161随后又证实了AlF3的催化性能要明显优于MgF2。目前这种金属氟化物催化体系的制备成本较高,但其突出的催化性能使得它们具有一定的应用潜力。
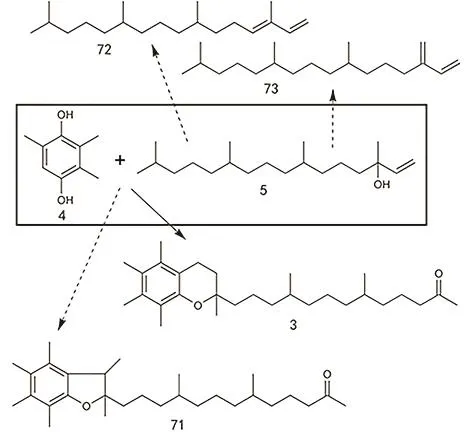
图30 TMHQ和异植物醇的缩合反应(实箭头代表主反应,虚箭头代表副反应)Fig. 30 Condensation of TMHQ and isophytol (solid arrow represent main reaction and dashed arrows denote side reactions).
含氟强酸是另一类研究较多的催化体系。它既可以是质子酸162,163,也可是其对应的金属盐164,165。该体系的一大特点是可以通过改变基团的种类来调控催化剂的酸性,具体可分为―OH、―NH、―CH三种类型的B酸,它们对应的金属盐又可表现出很强的L酸性。Bonrath等166,167发现―NH和―CH型B酸(图31)可以有效地催化TMHQ和异植物醇的缩合,产物的最高产率可达94.5%。随后他们又制备了上述几种B酸对应的金属盐,生育酚的产率保持在91%–94%之间168。Yamamoto等169对比了HOTf、HNTf2、C6F5CHTf2的B酸强弱,以及它们对应的三甲基硅化物的L酸强弱,得出B酸强弱顺序为HOTf > HNTf2> C6F5CHTf2,L酸强弱顺序为Me3Si[C6F5CHTf2] > Me3SiNTf2> Me3SiOTf,在催化活性方面,L酸和B酸的差别并不大,除Me3SiOTf外生育酚产率都在90%以上,但是由L酸催化副产物更少,得到的产品纯度更多,其中Me3Si[C6F5CHTf2]催化得到的产品纯度高达99.2%。Coman等170先制备了不同比例的Sn掺杂的介孔硅,随后将Tf基团吸附其上得到SnTf-MCM-41和SnTf-UVM-7两种催化剂,它们都表现出很强的L酸性,催化TMHQ和异植物醇缩合反应中TMHQ的转化率高达98%,生育酚选择性为94%,最重要的是它分离简单,性能较为稳定。含氟强酸类催化剂的活性和选择性都很高,较高的成本是其大规模应用的最大障碍。
图31 -NH型和-CH型含氟强酸的结构及制备方法167Fig. 31 Structure and preparation of perfluorinated imide and perfluorinated methide 167.
强酸性阳离子交换树脂在水解、酯化、缩合及烷基化反应中表现出优良的催化性能。而且催化剂易于回收利用,具有很广阔的应用前景。其中聚苯乙烯型Amberlyst 15和全氟磺酸树脂Nafion 50是最具代表性、应用最广泛的两类阳离子交换树脂。Bonrath等171将Amberlyst 15和Nafion 50应用于TMHQ和异植物醇的缩合反应中,生育酚的产率在75%–92%之间,溶剂的影响较为明显,在碳酸丙烯酯中产率最高,Amberlyst 15和Nafion 50分别为90%和92%。而且催化性能稳定,经历3个循环,产率几乎不变。随后他们又将Nafion 50负载于多孔二氧化硅上,这样可以很好地分散酸性位点,使得催化活性有所提升,采用碳酸丙烯酯为溶剂,异植物醇的转化率在95%以上,生育酚的产率为92%,而且催化活性与溶剂的极性紧密相连96。Wang等172制备了不同负载量(5%、13%、20% (w))Nafion/SiO2纳米复合材料,并将它们应用于TMHQ和异植物醇的缩合反应,发现使用5% (w)Nafion/SiO2时,异植物醇的转化率为98.4%,生育酚的选择性约为100%。随着负载量增加到20% (w)时,转化率和选择性都明显下降很多,而且选择性不到20%。分析表明负载后的Nafion催化剂酸位位点得以分散,所以催化剂活性会比Nafion本体高出一个数量级,负载后酸性的减弱有利于提高选择性,然而随着负载量增加,酸性逐渐增强,甚至恢复到Nafion本体的酸性,再辅以较高的传质,使得副反应大大加快,导致极低的生育酚选择性。
杂多酸和离子液体也被用于生育酚的合成。使用磷钨酸和硅钨酸时,产品中生育酚的纯度都可以达到90%以上173。Xing等174发现磺酸基功能化的离子液体可用于生育酚的制备,最高收率可达94.3%。离子液体最重要的一个特点是具有反应介质和催化剂的双重功能,而且反应较为绿色,但其作为一种新兴的催化剂,合成复杂,成本高。此外,硅铝分子筛兼备发达的孔道结构和较强的酸性175,在生育酚合成领域内也有着很大的应用前景。
除了催化剂外,溶剂也是生育酚合成体系至关重要的一环176。目前来看,在多数催化体系中,碳酸丙烯酯或者含有碳酸丙烯酯的混合溶剂是最佳的选择。近年来,对于超临界CO2以及离子液体的研究也取得不错的成果177,178。超临界CO2需要较为苛刻的反应条件,而且它对TMHQ的溶解度不高,因此应用受到了限制。
5 结论和展望
近年来,维生素E的市场需求急剧增加,合成维生素E占据市场总量的80%以上,开发合成维生素E的高效途径具有重大意义。维生素E主要由TMHQ和异植物醇缩合而来,该过程主要涉及酸催化的烷基化反应。TMHQ的主要由间甲苯酚先后经历邻位甲基化、氧化、加氢得来,近几年以丙酮缩合产物异氟尔酮为原料的合成路线受到广泛关注。异植物醇合成路线中的一个关键中间体是甲基庚烯酮,乙炔-丙酮法和丙酮-异丁烯-甲醛法是构筑甲基庚烯酮的常用方法,而炔醇选择性加氢是最为关键的一步,如何提高烯醇的选择性将是提升维生素E产量的重中之重。在开发维生素E合成路线的过程中应该根据当地原料供应情况、政府政策等合理选择最合适的工艺。
纵观整个合成工艺,有三点需要特别关注:(1)工艺中涉及多个酸催化的反应,目前来看质子酸催化活性很高,但难以分离,易腐蚀设备,造成环境污染,因此多项研究集中于固体酸催化剂的开发,但如何提高固体酸的活性和选择性以及固载酸催化剂的活性组分流失问题已经成为工作的重点;(2) 在TMHQ的制备过程中存在氧化反应,氧化反应的选择性最为关键,以氧气或空气为氧化剂,发展高效、高选择性、低污染的氧化催化剂是今后的主要发展方向;(3) 炔醇的选择性加氢是异植物醇合成中最为重要的一个反应,如何在保证高转化率的前提下提升烯醇的选择性尤为关键。
(1) Traber, M. G. Annu. Rev. Nutr. 2007, 27, 347.doi: 10.1146/annurev.nutr.27.061406.093819
(2) Sun, J. M.; Lu, H. F.; Nan, J. L. Tianjin Chem. Ind. 2002, 5, 26.[孙建梅, 陆洪芳, 南家莲. 天津化工, 2002, 5, 26.]doi: 10.3969/j.issn.1008-1267.2002.05.012
(3) Eckstein, S.; Hintermeier, P. H.; Olarte, M. V.; Liu, Y.; Baráth, E.;Lercher, J. A. J. Catal. 2017, 352, 329.doi: 10.1016/j.jcat.2017.06.002
(4) Leach B. L. Process for direct methylation of phenol in liquid phase.US 3994982A, 1975.
(5) Leach B. E. Process for the synthesis of 2,6-xylenol and 2,3,6-trimethylphenol. US 4283574A, 1980.
(6) Velu, S.; Sivasanker, S. Res. Chem. Intermediat 1998, 24, 657.doi: 10.1163/156856798x00555
(7) Grabowska, H.; Wrzyszcz, J.; Syper, L. Catal. Lett. 1999, 57, 135.doi: 10.1023/a:1019083112771
(8) Xiao, G. M.; Li, Z. S; Jiang, F.; Niu, L.; Xiong, Z. Catalyst for synthesizing 2,3,6-trimethylphenol and preparation method thereof.CN 102974354A, 2013.
(9) Niu, L.; Li, Z. S.; Jiang, F.; Zhou, M. H.; Wang, Z. H.; Xiao, G. M.React. Kinet. Mech. Cat. 2014, 112, 199.doi: 10.1007/s11144-014-0697-z
(10) Gandhe, A. R.; Fernandes, J. B. Catal. Commun. 2004, 5, 89.doi: 10.1016/j.catcom.2003.11.017
(11) Gandhe, A. R.; Naik, S. P.; Kakodkar, S. B.; Fernandes, J. B. Catal.Commun. 2006, 7, 285. doi: 10.1016/j.catcom.2005.09.013
(12) Santacesaria, E.; Grasso, D.; Gelosa, D.; Carrá, S. Appl. Catal. 1990,64, 83. doi: 10.1016/S0166-9834(00)81555-9
(13) Sreekumar, K.; Sugunan, S. Appl. Catal. A 2002, 230, 245.doi: 10.1016/S0926-860X(02)00006-6
(14) Bezouhanova, C.; Al-Zihari, M. A. Appl. Catal. A 1992, 83, 45.doi: 10.1016/0926-860X(92)80024-7
(15) Qian, D.; He, H. Q.; Wang, K. Y. Chem. Reagents (Beijing, China)2002, 4, 231. [钱东, 何厚群, 王开毅. 化学试剂, 2002, 4, 231.]doi: 10.3969/j.issn.0258-3283.2002.04.016.
(16) Bodnar, Z.; Mallat, T.; Baiker, A. J. Mol. Catal. A-Chem. 1996, 110,55. doi: 10.1016/1381-1169(96)00042-8
(17) Li, X.; Wang, X. Y.; Shu, W. G.; Qian, D. Hunan Chem. Ind. 1998,5, 24. [李雄, 王歆燕, 舒万艮, 钱东. 湖南化工, 1998, 5, 24.]doi: 10.19342/j.cnki.issn.1009-9212.1998.05.010
(18) Chu, Z. H.; Zang, H. C. Guangzhou Chem. Ind. 2011, 9, 104.[储振华, 臧恒昌. 广东化工, 2011, 9, 104.]doi: 10.3969/j.issn.1001-9677.2011.09.037
(19) Takehira, K.; Shimizu, M.; Watanabe, Y.; Orita, H.; Hayakawa, T.J. Chem. Soc., Chem. Commun. 1989, 1705.doi: 10.1039/C39890001705
(20) Takehira, K.; Shimizu, M.; Watanabe, Y.; Orita, H.; Hayakawa, T.Tetrahedron Lett. 1989, 30, 6691.doi: 10.1016/S0040-4039(00)70652-6
(21) Shimizu, M.; Watanabe, Y.; Orita, H.; Hayakawa, T.; Takehira, K.B. Chem. Soc. Jpn. 1992, 65, 1522. doi: 10.1246/bcsj.65.1522
(22) Sun, H. J.; Harms, K.; Sundermeyer, J. J. Am. Chem. Soc. 2004, 126,9550. doi: 10.1021/ja0391964
(23) Wang, X. P.; Yang, R. Y.; Li, W.; Li, X. A.; Yan, J.; Liu, W. T.;Zhang, H. H. Ind. Catal. 2013, 8, 73. [王宪沛, 杨瑞云, 李文, 李小安, 阎俊, 刘卫涛, 张辉辉. 工业催化, 2013, 8, 73.]doi: 10.3969/j.issn.1008-1143.2013.08.016
(24) Guan, W. H.; Wang, C. M.; Yun, X.; Hu, X. B.; Wang, Y.; Li, H. R.Catal. Commun. 2008, 9, 1979. doi: 10.1016/j.catcom.2008.03.028
(25) Wang, C.; Guan, W.; Xie, P.; Yun, X.; Li, H.; Hu, X.; Wang, Y.Catal. Commun. 2009, 10, 725. doi: 10.1016/j.catcom.2008.11.027
(26) Kholdeeva, O. A.; Golovin, A. V.; Kozhevnikov, I. V. React. Kinet.Catal. L. 1992, 46, 107. doi: 10.1007/bf02096685
(27) Kholdeeva, O. A.; Golovin, A. V.; Maksimovskaya, R. I.;Kozhevnikov, I. V. J. Mol. Catal. 1992, 75, 235.doi: 10.1016/0304-5102(92)80128-4
(28) Qian, D.; Zhang, M.; Wang, K. Y. Chin. J. Synth. Chem. 1998, 2, 2.[钱东, 张敏, 王开毅. 合成化学, 1998, 2, 2.]doi: 10.15952/j.cnki.cjsc.1998.02.001
(29) Palacio, M.; Villabrille, P. I.; Romanelli, G. P.; Vázquez, P. G.;Cáceres, C. V. Appl. Catal. A 2012, 417, 273.doi: 10.1016/j.apcata.2011.12.049
(30) Lin, T. H.; Chen, C. C.; Jang, L. Y.; Lee, J. F.; Cheng, S. Micropor.Mesopor. Mater. 2014, 198, 194.doi: 10.1016/j.micromeso.2014.07.027
(31) Trukhan, N. N.; Romannikov, V. N.; Paukshtis, E. A.; Shmakov, A.N.; Kholdeeva, O. A. J. Catal. 2001, 202, 110.doi: 10.1006/jcat.2001.3264
(32) Zhou, J.; Hua, Z. L.; Cui, X. Z.; Ye, Z. Q.; Cui, F. M.; Shi, J. L.Chem. Commun. 2010, 46, 4994. doi: 10.1039/C0CC00499E
(33) Kholdeeva, O. A.; Ivanchikova, I. D.; Guidotti, M.; Ravasio, N.;Sgobba, M.; Barmatova, M. V. Catal. Today 2009, 141, 330.doi: 10.1016/j.cattod.2008.06.005
(34) Wu, M. Z.; Li, Y.; Huang, X. Z.; Liu, W.; Yi, Z. Z. Appl. Chem. Ind.(Xi’an, China) 2014, 3, 456. [吴明珠, 李应, 黄相中, 刘卫,易中周. 应用化工, 2014, 3, 456.]doi: 10.16581/j.cnki.issn1671-3206.2014.03.004
(35) Li, Y.; Liu, W.; Wu, M. Z.; Yi, Z. Z.; Zhang, J. C. Mendeleev.Comm. 2010, 20, 218. doi: 10.1016/j.mencom.2010.06.012
(36) Liu, Y. C.; Hsu, J.; Fu, Y. P.; Tsai, K. Int. J. Hydrog. Energy 2016,41, 15696. doi: 10.1016/j.ijhydene.2016.04.127
(37) Zhang, T. Y.; Wang, M. Y.; Li, B.; Liu, Q. Chem. Ind. Eng. Prog.(Beijing, China) 2016, 2, 513. [张天永, 王梦颖, 李彬, 刘茜. 化工进展, 2016, 2, 513.] doi: 10.16085/j.issn.1000-6613.2016.02.025
(38) Zhu, Z. Q.; Yuan, Z. F.; Xu, Q. F. Preparation method of 2,3,5-trimethylhydroquinone. CN 102241577A, 2011.
(39) Zhang, T. Y.; Yin, G.; Li, B.; Wang, X.; Jiang, S.; Yuan, Z. F. Res.Chem. Intermediat 2015, 41, 663. doi: 10.1007/s11164-013-1219-8
(40) Qian, D.; Wang, K. Y.; Yang, L. Y.; Zhang, M. K. J. Cent. South Univ. (Sci. Technol.) 2000, 1, 41. [钱东, 王开毅, 杨礼义, 张茂昆.中南工业大学学报(自然科学版), 2000, 1, 41.]
(41) Qian, D.; Wang, K. Y.; Yang, L. Y.; Zhang, M. K. J. Cent. South Univ. (Sci. Technol.) 1998, 5, 97. [钱东, 王开毅, 杨礼义, 张茂昆.中南工业大学学报(自然科学版), 1998, 5, 97.]
(42) Zhao, X. M.; Jin, Y.; Zhang, F. M.; Zhong, Y. J.; Zhu, W. D. Chem.Eng. J. 2014, 239, 33. doi: 10.1016/j.cej.2013.11.003
(43) Zhang, T. Y.; Yin, G.; Li, B.; Deng, Y. F.; Yuan, Z. F. Appl. Chem.Ind. (Xi'an, China) 2013, 8, 1363. [张天永, 尹观, 李彬, 邓永峰,袁仲飞. 应用化工, 2013, 8, 1363.]doi: 10.16581/j.cnki.issn1671-3206.2013.08.034
(44) Mukhopadhyay, S.; Chandnani, K. H.; Chandalia, S. B. Org.Process. Res. Dev. 2000, 4, 254. doi: 10.1021/op990074z
(45) Su, D. F.; Wei, Z. Z.; Mao, S. J.; Wang, J.; Li, Y.; Li, H. R.; Chen, Z.R.; Wang, Y. Catal. Sci. Technol. 2016, 6, 4503.doi: 10.1039/C5CY02171E
(46) Qiao, J. C.; Chen, Q.; Cai, D. W. Process for synthesizing 2,3,6-trimethylphenol by 4-tert-butylphenol. CN 102976902A, 2013.
(47) Zhao, J. C.; Han, Q. Y.; Liu, J. F.; Liu, L.; Hu, L. L.; Liu, Y. H.Method for extraction of 2,4,6-trimethylphenol from 2,6-dimethylphenol waste. CN 104045522A, 2014.
(48) Teijin LTD Preparation of 2,4,6-trimethylphenol. GB 1451091A,1973.
(49) Bonrath, W.; Schuetz, J.; Cavani, F. Manufacture of 2,4,6-trimethylphenol. WO 2015197586A1, 2015.
(50) Ichikawa, Y.; Yamanaka, Y.; Tsuruta, H. Novel process for preparation of 4-hydroxy-2,4,6-trimethyl-2,5-cyclohexadiene-1-one.US 3966818A, 1973.
(51) Toyoda, Y.; Ikeda, Y.; Hase, T.; Kitano, N. Preparation of 4-hydroxy-2,4,6-trimethyl-2,5-cyclohexadien-1-one. JPS 5841835A,1981.
(52) Tomita, T.; Kino, M.; Takada, T. Preparation of 4-hydroxy-2,4,6-trimethylcyclohexa-2,5-dien-1-one. JPS 58116435A, 1981.
(53) Tomita, T.; Kino, M.; Takada, T. Preparation of hydroxy-tri:methylcyclo-hexa-dienone—by reacting tri:methyl-phenol with hypohalous acid. JPS 59163337A, 1983.
(54) Yoshida, D. Production of high-purity 4-hydroxy-2,4,6-trimethyl-2,5-cyclohexadien-1-one. JPS 62238230A, 1986.
(55) Gogou, T. Preparation of 4-hydroxy-2,4,6-trimethyl-2,5-cyclohexadien-1-one. JPS 5953438A, 1982.
(56) Costantini, M.; Igersheim, F.; Krumenacker, L. Process for the preparation of 4-hydroxy-2,4,6-trimethyl-2,5-cyclohexadienone. US 4565895A, 1984.
(57) Costantini, M.; Igersheim, F.; Krumenacker, L. 4-Hydroxy 2,4,6-tri:methyl cyclohexadienone preparation—by chlorination and hydrolysis of 2,4,6-tri:methyl phenol. US 4612401A, 1986.
(58) Teijin LTD Preparation of trimethyl hydroquinone. GB 1439494A,1973.
(59) Qiao, J. C.; Chen, Q.; Cai, D. W. Process for synthesizing 2,3,6-trimethylphenol by phenol. CN 102976903A, 2013.
(60) Arnold, L.; Pasedach, H.; Pommer, H. 2,3,6-Trimethylphenol production—by reacting diethyl ketone in presence of base with e.g.crotonaldehyde. DE 1668874B1, 1971.
(61) Tavs, P.; Laas, H.; Schauer, H.; Arnold, L. Increasing the yield of 2.5.6-trimethylcyclohex-2-en-1-one. US 4820874A, 1989.
(62) Rittinger, S.; Rieber, N.; Arnold, L.; Hoercher, U. Production of 2,3,6-tri:methylphenol—by reacting di:ethyl ketone with 1-aminovinyl methyl ketone at 50-200 deg.C. DE 4414877A1, 1995.
(63) Nissei IND CO LTD Trimethylbenzoquinone production in aq.soln.—from 3,6-dinitrotrimethyl benzene sulphonate using ferric salt as oxidising agent. JP 75028426B, 1975.
(64) Bao, J.; Liu, G. F.; Gao, R.; Zhang, Y. 2016, 4, 187. [包吉, 刘高赋,高荣, 张雨. 化工管理, 2016, 4, 187.] doi: 10.3969/j.issn.1008-4800.2016.04.121
(65) Chen, H.; Wu, Y.; Xu, G. M. Adv. Fine Petrochem. 2002, 4, 25.[陈红, 吴缨, 徐国梅. 精细石油化工进展, 2002, 4, 25.]doi: 10.3969/j.issn.1009-8348.2002.04.008
(66) Zhao, F.; Yang, B. Y.; Gu, J. J.; Guan, P. M. Ind. Catal. 2012, 6, 62.[赵峰, 杨蓓玉, 顾剑江, 管盘铭. 工业催化, 2012, 6, 62.]doi: 10.3969/j.issn.1008-1143.2012.06.014
(67) Zhang, T. Y.; Liu, X. S.; Li, B.; Wang, M. Y.; Wang, Z. C.; Hai, L.Chem. Bull. (Beijing, China) 2017, 6, 573. [张天永, 刘晓思, 李彬,王梦颖, 王智超, 海莉. 化学通报, 2017, 6, 573.]doi: 10.14159/j.cnki.0441-3776.2017.06.010
(68) Zhang, T. Y.; Duan, Y. J.; Li, B.; Wang, X.; Du, J.; Yin, G.; Yuan, Z.F. Chem. Reagents (Beijing, China) 2013, 1, 3. [张天永, 段永洁,李彬, 王晓, 杜晶, 尹观, 袁仲飞. 化学试剂, 2013, 1, 3.]doi: 10.13822/j.cnki.hxsj.2013.01.016
(69) Dawson, B.; Pugach, J. Method of making ketoisophorone via oxidation of isophorone with tert-butyl hydroperoxide. WO 9615094A1, 1996.
(70) Noesberger, P.; Vieth, A. Process for manufacture of betaisophorone. US 5276197A, 1994.
(71) Krill, S.; Giray, G.; Huthmacher, K.; Huebner, F.; Tanner, H.Method of producing 3,5,5-trimethylcyclohexa-3-ene-1-one( beta -isophorone) by the isomerization of 3,5,5-trimethylcyclohexa-2-ene-1-(alpha-isophorone). US 6005147A, 1999.
(72) Takahashi, I.; Shibata, H. Oxidation catalyst and oxidation process using the same. US 6462239B2, 2002.
(73) Ina, T.; Miura, H.; Takahashi, I. Process for the production of ketoisophorone derivatives and equipment therefor. US 6410797B1,2002.
(74) Murphy, E. F.; Baiker, A. J. Mol. Catal. A-Chem. 2002, 179, 233.doi: 10.1016/S1381-1169(01)00342-9
(75) Bellut, H. Method of producing 2,6,6-trimethyl-2-cyclohexane-1,4-dione. US 4970347A, 1990.
(76) Hu, X. B.; Mao, J. Y.; Sun, Y.; Chen, H.; Li, H. R. Catal. Commun.2009, 10, 1908. doi: 10.1016/j.catcom.2009.06.024
(77) Chen, Z. R.; Fang, T. T.; Yuan, S. F.; Yin, H. Int. J. Chem. Kinet.2016, 48, 295. doi: 10.1002/kin.20987
(78) Beyrhouty, M.; Sorokin, A. B.; Daniele, S.; Hubert-Pfalzgraf, L. G.New J. Chem. 2005, 29, 1245. doi: 10.1039/B507211E
(79) Sorokin, A. B.; Quignard, F.; Valentin, R.; Mangematin, S. Appl.Catal. A 2006, 309, 162. doi: 10.1016/j.apcata.2006.03.060
(80) Burns, E.; Huang, T.; Weare, W. W.; Bartolotti, L.; Wang, X. Y.;Yao, J.; Li, H. R.; Franzen, S. J. Mol. Catal. A-Chem. 2015, 410,110. doi: https://doi.org/10.1016/j.molcata.2015.09.015
(81) Thatte, C. S.; Rathnam, M. V.; Pise, A. C. J. Chem. Sci. 2014, 126,727. doi: 10.1007/s12039-014-0601-4
(82) Mao, J. Y.; Li, N.; Li, H. R.; Hu, X. B. J. Mol. Catal. A-Chem. 2006,258, 178. doi: 10.1016/j.molcata.2006.05.051
(83) Zhang, P. F.; Li, H. R.; Wang, Y. Chem. Commun. 2014, 50, 6312.doi: 10.1039/c4cc02676d
(84) Kishore, D.; Rodrigues, A. E. Catal. Commun. 2007, 8, 1156.doi: 10.1016/j.catcom.2006.10.037
(85) Kishore, D.; Rodrigues, A. E. Appl. Catal. A 2008, 345, 104.doi: 10.1016/j.apcata.2008.04.029
(86) Wang, C. M.; Wang, G. L.; Mao, J. Y.; Yao, Z.; Li, H. R. Catal.Commun. 2010, 11, 758. doi: 10.1016/j.catcom.2010.02.010
(87) Chen, K. X.; Sun, Y.; Wang, C. M.; Yao, J.; Chen, Z. R.; Li, H. R.Phys. Chem. Chem. Phys. 2012, 14, 12141.doi: 10.1039/C2CP41617D
(88) Zhong, W. Z.; Mao, L. Q.; Xu, Q.; Fu, Z. H.; Zou, G. Q.; Li, Y. Q.;Yin, D. L.; Luo, H.; Kirk, S. R. Appl. Catal. A 2014, 486, 193.doi: 10.1016/j.apcata.2014.08.005
(89) Murphy, E. F.; Mallat, T.; Baiker, A.; Schneider, M. Appl. Catal. A 2000, 197, 295. doi: 10.1016/S0926-860X(99)00491-3
(90) Crier, S.; Huthmacher, K. Method for preparation of 2,3,5-trimethyl hydrochinone di-ester. CN 1265390A, 2000.
(91) Weigel, H.; Krill, S.; Hasselbach, H.; Huthmacher, K.; Wegel, H.;Kelier, S.; Haserbach, H. Process for preparing esterified chroman compounds. US 6329535B1, 2001.
(92) Bonrath, W.; Schneider, M.; Werner, B.; Michael, S. Manufacture of trimethylhydroquinone diacylates. CN 1604888A, 2005.
(93) Weigel, H.; Krill, S.; Hasselbach, H.; Huthmacher, K.; Wegel, H.;Kelier, S.; Haserbach, H. Preparation process of exterified chroman compound. CN 1308077A, 2001.
(94) Wildermann, A.; Foricher, Y.; Netscher, T.; Bonrath, W. Pure Appl.Chem. 2007, 79, 1839. doi: 10.1351/pac200779111839
(95) Zeng, Q. Y.; Song, W. J.; Zhang, Q.; Pan, H.; Gao, J. Y.; Ni, C. Y.Method for synthesizing 2,3,5-trimethylhydroquinone diester. CN 102180793A, 2011.
(96) Schneider, M.; Zimmermann, K.; Aquino, F.; Bonrath, W. Appl.Catal. A 2001, 220, 51. doi: 10.1016/S0926-860X(01)00704-9
(97) Rác, B.; Molnár, Á.; Forgo, P.; Mohai, M.; Bertóti, I. J. Mol. Catal.A-Chem. 2006, 244, 46. doi: 10.1016/j.molcata.2005.08.043
(98) Hinze, R.; Laufer, M. C.; Hölderich, W. F.; Bonrath, W.; Netscher,T. Catal. Today 2009, 140, 105. doi: 10.1016/j.cattod.2008.07.008
(99) Aec Chim Organ Biolog. Improvements in and relating to process of preparing isophytol. GB 1087837A, 1967.
(100) Kuśtrowski, P.; Sułkowska, D.; Chmielarz, L.; Dziembaj, R. Appl.Catal. A 2006, 302, 317. doi: 10.1016/j.apcata.2006.02.003
(101) Abelló, S.; Vijaya-Shankar, D.; Pérez-Ramírez, J. Appl. Catal. A 2008, 342, 119. doi: 10.1016/j.apcata.2008.03.010
(102) Díez, V. K.; Di Cosimo, J. I.; Apesteguía, C. R. Appl. Catal. A 2008,345, 143. doi: 10.1016/j.apcata.2008.04.035
(103) Abelló, S.; Medina, F.; Tichit, D.; Pérez-Ramírez, J.; Rodríguez, X.;Sueiras, J. E.; Salagre, P.; Cesteros, Y. Appl. Catal. A 2005, 281,191. doi: 10.1016/j.apcata.2004.11.037
(104) Díez, V. K.; Apesteguía, C. R.; Di Cosimo, J. I. J. Catal. 2006, 240,235. doi: 10.1016/j.jcat.2006.04.003
(105) Hoelderich W.; Ritzerfeld, V. Preparing pseudoionone by aldol condensation of citral and acetone using heterogeneous catalyst,comprises utilizing supported heterogeneous catalysts based on rare earth metals applied on e.g. zirconium dioxide carrier as carrier materials. DE 102012012785A1, 2013.
(106) Wang, Z.; Lu, G. Z.; Guo, Y.; Guo, Y. L.; Gong, X. Q. ACS Sustain.Chem. Eng. 2016, 4, 1591. doi: 10.1021/acssuschemeng.5b01533
(107) Raju, V.; Radhakrishnan, R.; Jaenicke, S.; Chuah, G. K. Catal.Today 2011, 164, 139. doi: 10.1016/j.cattod.2010.10.043
(108) Horst, P.; Herbert, M.; Hermann, O. Preparing alkenones 2-methylheptene-1-on-6 from. DE 1268135B, 1968.
(109) Babler, J. H. Process for preparing tertiary alkynols. US 5349071A,1994.
(110) Vorobyeva, E.; Chen, Z.; Mitchell, S.; Leary, R. K.; Midgley, P.;Thomas, J. M.; Hauert, R.; Fako, E.; Lopez, N.; Perez-Ramirez, J.J. Mater. Chem. A 2017, 5, 16393. doi: 10.1039/C7TA04607C
(111) Crespo-Quesada, M.; Yarulin, A.; Jin, M.; Xia, Y.; Kiwi-Minsker, L.J. Am. Chem. Soc. 2011, 133, 12787. doi: 10.1021/ja204557m
(112) Yarulin, A.; Yuranov, I.; Cárdenas-Lizana, F.; Abdulkin, P.;Kiwi-Minsker, L. J. Phys. Chem. C 2013, 117, 13424.doi: 10.1021/jp402258s
(113) Semagina, N.; Renken, A.; Kiwi-Minsker, L. J. Phys. Chem. C 2007,111, 13933. doi: 10.1021/jp073944k
(114) Tripathi, B.; Paniwnyk, L.; Cherkasov, N.; Ibhadon, A. O.; Lana-Villarreal, T.; Gómez, R. Ultrason. Sonochem. 2015, 26, 445.doi: 10.1016/j.ultsonch.2015.03.006
(115) Tschan, R.; Schubert, M. M.; Baiker, A.; Bonrath, W.; Lansink-Rotgerink, H. Catal. Lett. 2001, 75, 31.doi: 10.1023/a:1016727904935
(116) Tschan, R.; Wandeler, R.; Schneider, M. S.; Burgener, M.; Schubert,M. M.; Baiker, A. Appl. Catal. A 2002, 223, 173.doi: 10.1016/S0926-860X(01)00755-4
(117) Studt, F.; Abild-Pedersen, F.; Bligaard, T.; Sørensen, R. Z.;Christensen, C. H.; Nørskov, J. K. Science 2008, 320, 1320.doi: 10.1126/science.1156660
(118) Yarulin, A.; Yuranov, I.; Cárdenas-Lizana, F.; Alexander, D. T. L.;Kiwi-Minsker, L. Appl. Catal. A 2014, 478, 186.doi: 10.1016/j.apcata.2014.04.003
(119) SNAM SPA(SNAM-C). Selective hydrogenation of alkynols to alkenols—in aqs medium using palladium catalyst and soluble zinc cpd. NL 136588B, 1972.
(120) Okhlopkova, L. B.; Matus, E. V.; Prosvirin, I. P.; Kerzhentsev, M.A.; Ismagilov, Z. R. J. Nanopart. Res. 2015, 17, 475.doi: 10.1007/s11051-015-3289-6
(121) Deng, D. S.; Yang, Y.; Gong, Y. T.; Li, Y.; Xu, X.; Wang, Y.Green Chem. 2013, 15, 2525. doi: 10.1039/c3gc40779a
(122) Shen, L. F.; Mao, S. J.; Li, J. Q.; Li, M. M.; Chen, P.; Li, H. R.;Chen, Z. R.; Wang, Y. J. Catal. 2017, 350, 13.doi: 10.1016/j.jcat.2017.01.021
(123) Bonrath, W.; Mueller, T.; Kiwi-Minsker, L.; Renken, A.; Iouranov,I.; Kiwi, M. Hydrogenation process. CN 102741206A, 2012.
(124) Bonrath, W.; Kiwi-Minsker, L.; Iouranov, I.; Kiwi, M. New catalytic system. CN 103906569A, 2014.
(125) Bonrath, W.; Buss, A. Metal powderdous catalyst comprising a Fealloy. CN 104136114A, 2014.
(126) Grjaznov, V.; Keravanov, A.; Belosljudo, N.; Ermolaev, A.;Maganjuk, A.; Saryceva, I. Process for the preparation of ethylene alcohols having 4 to 10 carbon atoms. DE 3114240A1, 1982.
(127) Kido, Y.; Kumagai, N.; Iwasaki, H.; Onishi, T.; Ueyama, F.;Kamiyama, F.; Kajiyashiki, T.; Kito, Y.; Iwagasaki, S. Process for producing 6-methyl-3-hepten-2-one and 6-methyl-2-heptanone analogues, and process for producing phyton or isophytol. US 5955636A, 1999.
(128) Wilson, S. R.; Price, M. F. J. Org. Chem. 1984, 49, 722.doi: 10.1021/jo00178a036
(129) Constant, S.; Tortoioli, S.; Müller, J.; Lacour, J. Angew. Chem. Int.Ed. 2007, 46, 2082. doi: 10.1002/anie.200604573
(130) Linder, D.; Buron, F.; Constant, S.; Lacour, J. Eur. J. Org. Chem.2008, 5778. doi: 10.1002/ejoc.200800854
(131) Linder, D.; Austeri, M.; Lacour, J. Org. Biomol. Chem. 2009, 7,4057. doi: 10.1039/B910475E
(132) Bizet, V.; Lefebvre, V.; Baudoux, J.; Lasne, M.-C.; Boulangé, A.;Leleu, S.; Franck, X.; Rouden, J. Eur. J. Org. Chem. 2011, 4170.doi: 10.1002/ejoc.201100120
(133) De Castro, K. A.; Byun, E. Y.; Rhee, H. B. Kor. Chem. Soc. 2009,30, 2155. doi: 10.5012/bkcs.2009.30.9.2155
(134) Oost, C.; Stroezel, M.; Etzrodt, H.; Weller, D.; Rheude, U.; Kaibel,G.; Krug, T.; Spiske, L.; Jaedicke, H.; Dietmar, W.; et al.Preparation of higher unsaturated ketone. CN 1271716A, 2000.
(135) Oost, C.; Stroezel, M.; Etzrodt, H.; Weller, D.; Bockstiegel, B.;Reimer, K.; Kaibel, G.; Jaedicke, H.; Dietmar, W.; Aust, C.; et al.Method for continuous preparation of unsaturated ketone. CN 1251832A, 2000.
(136) Takasago Perfumery CO (TAKS-C). Soprene trimer preparation—using a catalyst contg. zero-valent nickel and phosphines. JP 77016084B, 1977.
(137) Akutagawa, S. Geranyl acetone preparation—by reacting myrcene with acetone imine in presence of alkali metal catalyst, and hydrolysing. JP 53071005A, 1978.
(138) Kido, Y.; Kitayama, M.; Yoneda, K.; Iwasaki, H.; Onishi, T.;Kitayama, K. Process for producing 6-methylheptan-2-one. US 5840992A, 1998.
(139) Fujita, Y.; Wada, T.; Onishi, T.; Nishida, T.; Omura, S.; Mori, F.;Hosogai, T.; Aihara, F. 6,10-Di:methyl-6,9-undeca:diene-2-one production—by ethynylating substd. ketone obtd. from prenyl halide and mesityl oxide, partially hydrogenating and thermally rearranging. JP 55055131A, 1980.
(140) Kuraray Co LTD(KURS-C). Preparation of phytone and isophytol.JPS 53105409A, 1978.
(141) Onishi, T.; Fujita, Y.; Nishida, T.; Ishiguro, M.; Hosogai, T. Phytone or isophytol production—by heating propargyl alcohol cpd.hydrogenating the resulting ketone and opt. vinylation of resulting phytone. JP 54014906A, 1979.
(142) De Jong, A. J.; Van, H. R. Process for the preparation of 6,10,14-trimethylpentadecan-2-one. CH 623021A5, 1981.
(143) Kuraray Co LTD(KURS-C). 3, 7-dimethyl-1-octanal preparation—by hydroformylation of 2,6-dimethyl-2-or -3-heptenes. JP 74020170,1974.
(144) Goebbel, H.; Kaibel, G.; Miller, C.; Dobler, W.; Dirnsteiner, T.;Hahn, T.; Breuer, K.; Aquila, W.; Gobbel, H.; Gobbel, G.; et al.Production of tetrahydrogeranylacetone, for use in production of Vitamins E and K, involves selective, liquid-phase hydrogenation of pseudo-ionone using a device which inhibits the transport of catalyst particles. CN 1668564A, 2005.
(145) Clamoe, A.; Siegel, W. Method for preparing higher ketone by unsaturated aldehyde. CN 1330062A, 2002.
(146) Teles, J.; Hoffmann, W. Preparation of hexahydrofarnesylacetone form 6, 7-dihydrogeraniol, and novel intermadiates therefor.CN 1172795A, 1998.
(147) Zhao, Z. D.; Liu, X. Z. Biomass Chem. Eng. 2001, 2, 41. [赵振东,刘先章. 林产化工通讯, 2001, 2, 41.]doi: 10.3969/j.issn.1673-5854.2001.02.011
(148) Luo, J. Y.; Wang, H. Z.; Peng, S. J. Chem. Ind. For. Prod. 2000, 3,47. [罗金岳, 王汉忠, 彭淑静. 林产化学与工业, 2000, 3, 47.]doi: 10.3321/j.issn:0253-2417.2000.03.009
(149) Liu, X. Z.; Hu, X. E.; Jiang, T. F.; Li, D. M.; Hu, G. X. Chem. Ind.For. Prod. 1997, 3, 25. [刘先章, 胡樨萼, 蒋同夫, 李冬梅, 胡贵贤. 林产化学与工业, 1997, 3, 25.]doi: 10.3321/j.issn:0253-2417.1997.03.005
(150) Sun, M. H.; Qi, X.; Tang, X. Y.; Zhao, W. T. Chem. Ind. Eng.(Tianjin, China) 2016, 1, 51. [孙美环, 齐欣, 唐向阳, 赵温涛.化学工业与工程, 2016, 1, 51-56.]doi: 10.13353/j.issn.1004.9533.20131188
(151) Renninger, N. S.; Newman, J.; Reiling, K. K.; Regentin, R.; Paddon,C. J. Production of isoprenoids. WO 2007140339A2 2007.
(152) Renninger, N. S.; Mcphee, D. J. Fuel compositions comprising farnesane and farnesane derivatives and method of making and using same. US 2008083158A1, 2008.
(153) Tsuruta, H.; Lenihan, J. R.; Regentin, R. WO 2009042070A2, 2009.
(154) Chua, P. R.; Meadows, A. Methods for stabilizing production of acetyl-coenzyme a derived compounds. WO 2015020649A1, 2015.
(155) Lowack, R.; Meyer, J.; Eggersdorfer, M.; Grafen, P. Preparation of alpha-tocopherol and alpha-tocopheryl acetate in liquid or supercritical carbon dioxide. US 5523420A, 1996.
(156) Baldenius, K.; Kaiser, W.; Bockstiegel, B.; Laas, H.; Schulz, B.;Schmitt, P.; Glietenberg, H. Preparation of alpha-tocopherol or alpha-tocopheryl acetate by reacting trimethylhydroquinone and phytol or isophytol, with recycling of the zinc halide condensation catalyst. US 6005122A, 1999.
(157) Krill, S.; Kretz, S.; Huthmacher, K. Process for the production of alpha-tocopherol acetate by condensation of trimethylhydroquinone with isophytol. EP 1132384A2, 2001.
(158) Duan, H. Y.; Wang, Z. H.; Li, J. T.; Li, S. H.; Li, L. J.; Li, T. S.Synthetic. Commun. 2003, 33, 1867. doi: 10.1081/SCC-120020197
(159) Coman, S. M.; Wuttke, S.; Vimont, A.; Daturi, M.; Kemnitz, E.Adv. Synth. Catal. 2008, 350, 2517. doi: 10.1002/adsc.200800411
(160) Wuttke, S.; Coman, S. M.; Scholz, G.; Kirmse, H.; Vimont, A.;Daturi, M.; Schroeder, S. L. M.; Kemnitz, E. Chem. -Eur. J. 2008,14, 11488. doi: 10.1002/chem.200801702
(161) Candu, N.; Wuttke, S.; Kemnitz, E.; Coman, S. M.; Parvulescu, V. I.Appl. Catal. A 2011, 391, 169. doi: 10.1016/j.apcata.2010.08.004
(162) Baak, M.; Bonrath, W.; Pauling, H. Process for manufacturing d,1-alpha-tocopherol. CN 1237163A, 1999.
(163) Bonrath, W.; Gockel, S.; Haas, A.; Netscher, T.; Pauling, H.Preparation of (all-rac)-alpha-tocopherol, which is an important member of vitamin E group, involves acid catalysis of trimethylhydroquinone with isophytol or phytol in presence of bis(perfluoroalkylsulfonyl)-methane. WO 2003070718P1, 2003.
(164) Bonrath, W.; Wang, S. N. Preparation of d,1-alpha-tocopherol.US 6423851B2, 2002.
(165) Bonrath, W.; Dittel, C.; Netscher, T.; Pabst, T.; Giraudi, L. Process for the manufacture of alpha-tocopheryl acetate. WO 2004063182A1, 2004.
(166) Bonrath, W.; Haas, A.; Hoppmann, E.; Netscher, T.; Pauling, H.;Schager, F.; Wildermann, A. Adv. Synth. Catal. 2002, 344, 37.doi: 10.1002/1615-4169(200201)344:1<37::aid-adsc37>3.0.co;2-4
(167) Netscher, T.; Bonrath, W.; Haas, A.; Hoppmann, E.; Pauling, H.Chimia Inter. J. Chem. 2004, 58, 153.doi: 10.2533/000942904777678181
(168) Bonrath, W.; Dittel, C.; Giraudi, L.; Netscher, T.; Pabst, T.Catal. Today 2007, 121, 65. doi: 10.1016/j.cattod.2006.11.022
(169) Hasegawa, A.; Ishihara, K.; Yamamoto, H. Angew. Chem. Int. Ed.2003, 42, 5731. doi: 10.1002/anie.200352382
(170) Coman, S. M.; Pop, G.; Stere, C.; Parvulescu, V. I.; El Haskouri, J.;Beltrán, D.; Amorós, P. J. Catal. 2007, 251, 388.doi: 10.1016/j.jcat.2007.08.001
(171) Schager, F.; Bonrath, W. J. Catal. 1999, 182, 282.doi: 10.1006/jcat.1998.2351
(172) Wang, H.; Xu, B. Q. Appl. Catal. A 2004, 275, 247.doi: 10.1016/j.apcata.2004.07.038
(173) Kozhevnikov, I. V.; Kulikov, S. M.; Chukaeva, N. G.; Kirsanov, A.T.; Letunova, A. B.; Blinova, V. I. React. Kinet. Catal. L. 1992, 47,59. doi: 10.1007/bf02063560
(174) Xing, H. B.; Wang, T.; Zhou, Z. H.; Dai, Y. Y. Synthetic. Commun.2006, 36, 2433. doi: 10.1080/00397910600781166
(175) Laha, S. C.; Venkatesan, C.; Sakthivel, A.; Komura, K.; Kim, T. H.;Cho, S. J.; Huang, S. J.; Wu, P. H.; Liu, S. B.; Sasaki, Y.; et al.Micropor. Mesopor. Mater. 2010, 133, 82.doi: 10.1016/j.micromeso.2010.04.018
(176) Hirose, N.; Inoue, H.; Matsunami, T.; Yoshimura, T.; Morita, K.;Horikawa, Y.; Iwata, N.; Hayashi, K.; Seki, C.; Minami, N.; et al.Process for the preparation of alpha-tocopherol. CN 1123278A,1996.
(177) Kokubo, Y.; Hasegawa, A.; Kuwata, S.; Ishihara, K.; Yamamoto,H.; Ikariya, T. Adv. Synth. Catal. 2005, 347, 220.doi: 10.1002/adsc.200404312
(178) Xing, H. B.; Wang, T.; Dai, Y. Y. J. Supercrit. Fluid. 2009, 49, 52.doi: 10.1016/j.supflu.2008.12.003
