Advances in Electrode Materials for Aqueous Rechargeable Sodium-Ion Batteries
2018-07-03LIUShuangSHAOLianyiZHANGXuejingTAOZhanliangCHENJun
LIU Shuang, SHAO Lianyi, ZHANG Xuejing, TAO Zhanliang, CHEN Jun
Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Collaborative Innovation Center of Chemical Science and Engineering, College of Chemistry, Nankai University, Tianjin 300071, P. R. China.
1 引言
随着可再生能源和清洁能源的发展,产生了大规模储能的需求1。为了顺利接入、充分消纳可再生能源发电,实现能源的优化管理和高效利用,需要集中式储能、分布式储能、直流配电网的协调应用。虽然储能可以利用机械能、化学能、电磁场等多种形式,而电化学储能由于使用方便,节能减排,绿色低碳且经济仍然是今后大规模储能的发展方向,同时二次电池也因其安全性、经济性、模块化、维护方便在电化学储能技术中脱颖而出2。原则上,适合于大规模储能应用的二次电池须安全性高,环境友好,成本低廉,资源丰富并且具有优良的电化学性能如长寿命,高功率密度等。目前,电化学储能电站遵照 GB 51048-2014《电化学储能电站设计规范》,可采用铅酸电池、钠硫电池、全钒液流电池、锂离子电池等多种蓄电池,但上述电池尚不能满足大规模储能需求。铅酸电池成本低、运行可靠,但使用重金属铅,其能量密度和使用寿命不能满足储能需要;钠硫电池在储能领域具有理想的能量密度和功率密度,但需要在高温下工作,易发生燃烧事故;全钒液流电池使用寿命长,但能量密度偏低,关键材料受价格因素制约;锂离子电池在电动汽车动力电源领域展现了良好的发展前景,但单体容量较小,大量单体使用时电池管理技术复杂。同时,地壳中锂的相对丰度低(低于 20 ×10−6)、分布不均匀以及日益增长的价格使其难以满足大规模储能需求3–6,此外这些电池所采用的电解液也具有一定的毒性和腐蚀性7,8。
与锂相比,钠资源在地壳中分布广泛,丰度高,具有与锂相似的物理化学性质9(表1)和工作原理(图1),即均为“摇椅式”二次电池工作原理。充电时,钠离子从正极富钠材料中脱出进入溶液,同时溶液中钠离子嵌入负极材料;放电过程正好与之相反。在资源和环境等因素方面,钠离子电池作为储能应用具有很大优势。
钠离子电池并非一种新型的化学电源体系,早在20世纪70–80年代,钠离子电池的发展几乎与锂离子电池同时起步,随着索尼公司实现锂离子电池的商业化,钠离子电池的研究被逐渐忽视。这主要是由于钠离子质量较重且半径(0.102 nm)比锂离子(0.069 nm)大9,在充放电过程中,钠离子在电极材料中脱嵌困难、缓慢,主体材料膨胀/收缩引起的形变更容易使电极材料发生较大的应力变化,晶体结构发生瓦解,导致材料的循环稳定性较差。因此,不能将锂离子电池中成功应用的材料简单地移植到钠离子电池体系,开发能够高效、稳定储钠的电极材料成为当下研究热点。而对于大多数固定式储能场合,基于成本和安全性,人们将钠离子电池的研究拓展到水溶液电解质体系。

表1 金属钠与锂物理化学性质、分布及成本对比9Table 1 The comparison between Na and Li elements9.

图1 水溶液钠离子电池的反应原理示意图Fig. 1 Schematic illustration of the working mechanism of aqueous sodium ion battery.
水溶液电解质体系具有以下特点:(1) 水溶液电解液代替有机电解液,解决了易燃等安全性问题;(2) 生产条件相对宽松,电解液溶剂和盐价格相对便宜;(3) 钠离子水合离子半径(0.358 nm)比锂离子较小(0.382 nm),移动速率更快;(4) 水溶液离子电导率比有机电解液高 2个数量级,即使大尺寸的电极也能实现较高效率和能量密度10,11。因此水系钠离子电池被认为是最有潜力的大规模储能系统电池之一12。
在水系电解液中,钠离子电池的反应热力学性质受到水分解反应的严重影响,存在着水分解引起的负极析氢和正极析氧的副反应问题。另外,许多钠盐化合物在水中的溶解度很大,或遇水容易分解,进一步限制了储能材料的选择范围。基于此,电池材料应当满足以下条件13,14:(1) 氧化还原电对位于析氢析氧电位之间(如图 2);(2) 较高的电位下,溶液中的O2可以与电极材料发生副反应Na-(intercalated) + 1/2H2O + 1/4O2→ Na++OH–15,因此要求电极材料在操作pH下具有一定化学稳定性,不发生电极材料的溶解,并且不与氧发生反应;(3) 正极材料中应含有Na+,或者负极材料预嵌钠;(4) 可逆性好;(5) 安全。不同的电极材料已经被发展适用于水系钠离子电池。如图2,正极材料主要包括氧化物、聚阴离子材料以及普鲁士蓝类似物等;负极材料主要包括金属氧化物,NASICON型化合物,普鲁士蓝类似物以及有机化合物等。
2 水系钠离子电池电极材料研究现状
2.1 正极材料
2.1.1 过渡金属氧化物
在众多电化学储能材料中,过渡金属氧化物由于成本低、易合成、电化学活性高和稳定性好及形貌可控等优点受到越来越多的关注。之前的研究表明,过渡金属氧化物(如MnO2,RuO2,V2O5等)具有较好的法拉第过程,在超级电容器上得到普遍应用与研究。Ghodbane等16合成了一系列隧道结构MnOOM (M代表碱金属离子如Na+),发现一些大尺寸型隧道或层状结构的电容可达 150–300 F∙g–1,通过进一步实验,他们发现其储能机制为 Na+在其中嵌入/脱出,而不是表面电容效应,证明了嵌钠的可能。
2.1.1.1 二氧化锰(MnO2)
MnO2晶胞为一个锰原子被六个氧原子包围形成八面体,通过共用顶点位置和边缘的棱而形成隧道晶型结构,通过控制合成路线可得到不同隧道晶型结构的MnO2,一般主要包括α、β、γ、δ、λ五种晶型结构。根据隧道结构的不同可将MnO2分成3类:α、β、γ三种晶型结构具有一维隧道结构,δ-MnO2为二维层状结构17,18,λ-MnO2具有三维尖晶石结构19。

图2 水系钠离子电池电极材料在水溶液中电位(vs SHE, vs Na+/Na)13Fig. 2 The potentials of the electrode materials are described vs SHE, Na+/Na 13.
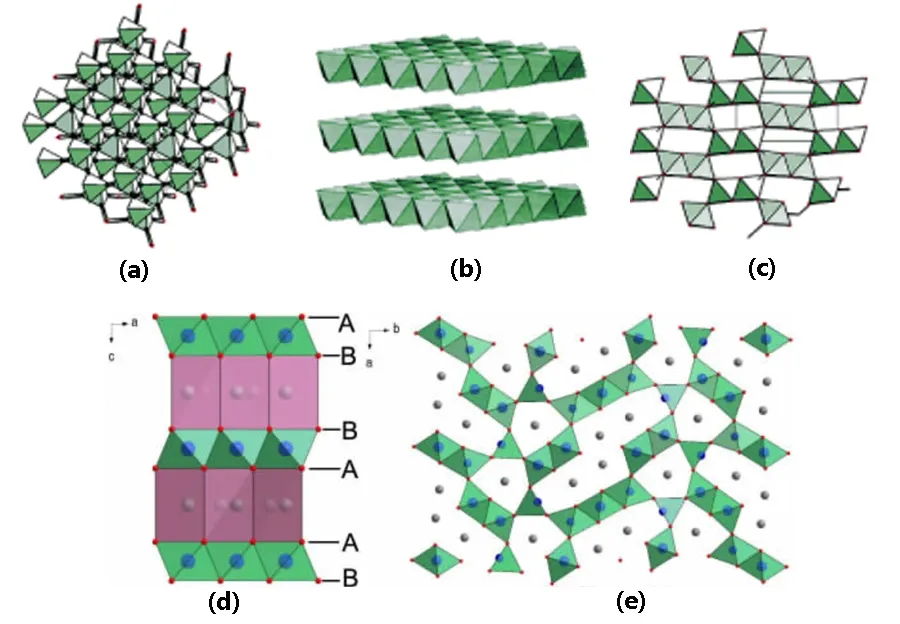
图 3 (a) λ-MnO2,(b) δ-MnO2,(c) γ-MnO2,(d) P2相-Na x MnO2,(e) P3相-Na x MnO2锰氧化物的结构示意图31Fig. 3 Structural illustrations of (a) λ-MnO2,(b) δ-MnO2, (c) γ-MnO2, (d) P2-type Na x MnO2 and(e) P3-type Na x MnO2 31.
Tarascon 等20首先报道了在有机电解质体系中尖晶石型 λ-MnO2(图 3a)中的钠离子嵌入/脱出行为。他们发现,Na嵌入到 λ-MnO2中会导致不可逆相变形成NaxMnO2结构,可逆嵌/脱钠离子数量为0.6。δ-MnO2(图3b)经常以水合物形式出现,可表示为 AxMnO2·n H2O (A = K+或 Na+)。最初对于 δ-MnO2的研究主要集中在超级电容器21–23。Kanoh与Shao等21,24通过非原位XRD分析证实了δ-MnO2在NaCl和Na2SO4水溶液电解液中钠离子的可逆嵌入和脱出。100 mA∙g−1下,电容量为142 F∙g−1,4000 mA∙g−1高电流密度下,也可保持其容量的80%,并可稳定循环850圈。2008年,Komaba等25发现钠离子可以在层状水钠锰矿(birnessite)型MnO2结构间可逆的嵌入/脱出,在水溶液中的可逆容量为 210 F∙g–1。
Minakshi等26报道了 γ-MnO2(图 3c)作为ARSB中正极材料的使用。使用7 mol∙L−1NaOH水溶液为电解液,在8 mA∙g−1的电流密度下,尽管比容量可达到225 mAh∙g−1,但是随着电化学反应的进行,仅循环25圈便有24%的容量衰减。
此外,人们还研究了其他过渡金属氧化物作为水溶液嵌钠正极的性能,如以V2O5作为ARSB正极材料,在电流密度为100 mA∙g−1时,可逆容量为43 mAh∙g−127。但其充放电曲线多表现为多级电压台阶,循环稳定性较差,应用价值不高。产生这一差别可能是由于体积较大的钠离子在氧化物晶格中比较稳定,可嵌脱的数量有限,且嵌脱反应容易引起结构变化。
2.1.1.2 NaxMnO2
钠基层状过渡金属氧化物NaxMO2(M = Co、Mn、Fe、Cr、Ni等)是一种研究较多的钠离子电池正极材料,具有多种晶体形态、结构和物理性能28。其中 NaxMnO2:0.45 ≤ x ≤ 0.7 时,为 P2 相-层状结构(图 3d);x < 0.45 时为 P3 相-三维隧道结构(图3e)。一般来说,层状 NaxMnO2能释放较高的实际容量,但充放电过程中产生的形变应力会造成结构坍塌和无定型化,导致循环性能不佳29,30。P3相-Na0.44MnO2由于具有较高的比容量和循环稳定性而被广泛研究,其具有三维互通的钠离子 S型隧道结构,包含大量的 c轴空缺,使钠离子能够快速扩散31。
Na0.44MnO2的隧道结构,早在1971年就被提出32,近年来得到了广泛研究。第一性原理揭示了 NaxMnO2的七个中间相反应(0.19 < x < 0.66)33,由于电压窗口的限制,Na0.44MnO2参与反应的部分为 0.22 < x < 0.44。Kim 等34通过 EIS 阻抗谱探究了Na0.44MnO2中钠离子扩散过程,测出离子扩散速率为 1.08 × 10−13–9.15 × 10−12cm2∙s−1,远远高于非水溶剂中的值,因此在水系电解液中,可实现大倍率充放电。此后,人们进一步采用多种方法合成以提高其性能,如固相法,水热法,溶胶凝胶法等。Liu等35通过 sol-gel法合成了棒状Na0.44MnO2,容量为 55 mAh∙g−1(200 F∙g−1),500 mA∙g−1(18C)下循环 4000圈后,容量保持率为84%。Dai等36通过聚乙烯吡咯烷酮燃烧法,在900 °C下制备的Na0.44MnO2具有快速的钠离子扩散速率和循环稳定性。C/5下,容量达 122.9 mAh∙g−1,在 20C 下,容量也可保持 99 mAh∙g−1。10C下,循环700圈后,容量保持率为82.9%。
Zhang等37比较了 Na0.35MnO2和 Na0.95MnO2的电化学性能,发现 Na0.35MnO2在 200 mA∙g−1下,可逆容量达40 mAh∙g−1,可稳定循环5000圈,具有更好的电化学性能。Tever和Whitacre38通过调整Na : Mn前驱体的比例,确定0.55为最稳定的电化学活性比例,其中包含微量的 β-Na0.7MnO2和 α-Mn2O3,C/1.4下循环 20圈后,可得到 35 mAh∙g−1比容量。最近,钱逸泰课题组39通过沉淀法合成了水钠锰矿型 Na0.58MnO2·0.48H2O,并用作水系钠离子电池正极,具有好的循环性能与倍率性能。1C 倍率下,可逆容量在 80 mAh∙g−1。10C倍率下,循环1000圈后,容量未见衰减,在50C的高倍率下,容量为54 mAh∙g−1。Yu等40通过固态法合成了 P2型-Na2/3Ni1/4Mn3/4O2,并以活性炭为负极探究了其在Li+/Na+混合水溶液电解液中的电化学性能,电池电压为 1.2 V,功率密度为 16 W∙kg−1时,能量密度为 36 Wh∙kg−1(以正负极活性材料总量计),但是循环稳定性较差。除NaxMnO2外,Liu等41,42也合成了KxMnO2并用于水系钠离子电池正极材料。
为了提高层状正极材料的结构稳定性和电化学性能,人们常采用阳离子取代的方法。例如,Wang等43通过固态法合成了 Ti取代的Na0.66[Mn0.66Ti0.34]O2,具有与 Na0.44MnO2相同的隧道结构,2C下,可逆容量为76 mAh∙g−1,高于Na0.44[Mn0.44Ti0.56]O2(45 mAh∙g−1)的容量。此外,Jung等44采用固态法合成了与Na0.44MnO2具有相同结构的Na2.7Ru4O9,由于钠离子在脱嵌后可重新嵌入的数量少于 60%,可逆容量较低。并且在 3 mA∙g–1的电流密度下只能循环几圈,这可能与溶剂化作用如质子共嵌入等有关。
2.1.2 聚阴离子化合物
聚阴离子型化合物是一系列含有四面体或者八面体阴离子结构单元(XOm)n−(X = P、S、As、Mo、W)的化合物的总称,其中MO6(M为过渡金属)八面体和XO4(X = P、S、As等) 四面体通过共顶点或者共边的方式连接形成三维网状结构,形成可被其他金属离子占据的空隙。结构稳定45、放电电位平台可调是该类材料的特点46,但是其电子电导率低47。因此需要通过碳包覆等改性方法使其能够达到实用水平。
2.1.2.1 NASICON-型
钠离子快导体结构(NASICON)结构可表示为NaxM2(PO4)3(M = 金属),其中金属占据与六个氧原子配位的八面体空隙,磷占据与四个氧原子配位的四面体空隙。NASICON类材料一般含有无毒且地球储量丰富的元素,如Fe等。
Na3V2(PO4)3作为一类钠离子超导体具有很强的离子扩散速率(图4a),可在3.4 V (vs Na/Na+)电压下发生两个钠离子的脱嵌,产生 117 mAh∙g−1的比容量48。Song等49探究了 NASICON型Na3V2(PO4)3在不同水溶液电解液如 1 mol∙L−1Li2SO4,Na2SO4和 K2SO4中的电化学行为,发现在Na2SO4电解液中,8.5C下,比容量为209 F∙g−1(~50 mAh∙g−1),42.7C 下,比容量为 136 F∙g−1(~35 mAh∙g−1)。不同扫速的循环伏安实验揭示了Na3V2(PO4)3电极在水溶液中的嵌/脱钠速率是由扩散控制的。并且其氧化还原电位为~0.4 V (vs SCE),8.5C下可循环30圈。将其中一半的V替换为Ti,可得到Na2VTi(PO4)3。如Mason和Lange50通过Ti替换V制备了Na2VTi(PO4)3,由于Ti4+是不溶的,当被水性电解质侵蚀时,可钝化形成不同水合的氧化物,降低了Na3V2(PO4)3做电极的副反应,表现出高水平稳定性。
NASICON结构电极材料具有良好的动力学和循环稳定性,但钒源中五价钒毒性较大,在一定程度上限制了其大规模使用,因此开发使用无毒的,含量丰富的元素(如Fe、Mn、Ni等)来制备新型 NASICON结构化合物,将是一个有意义的研究方向。
2.1.2.2 橄榄石结构
Fernandez-Ropero等51制备了橄榄石型NaFePO4(图 4b),与有机电解液相比,NaFePO4在室温下与 55 °C条件下均表现出较低的极性以及较好的倍率性能。在室温条件下,2C倍率下比容量为 70 mAh∙g−1,55 °C,C/5 下比容量达 110 mAh∙g−1。Vujkovic 和 Mentus52证明了橄榄石型LiFePO4能够在 NaNO3水溶液电解液中可完全转变为NaFePO4进行可逆循环,与有机电解液中相比,具有更好的倍率性能。Levi和 Zhao等53,54研究了LixFePO4在含Li+、Na+的混合电解液中的电化学行为,发现Na+也能在LixFePO4中可逆嵌/脱,过程中产生的中间相Na0.7FePO4,与Moreau在有机体系中的报道一致55。

图4 聚阴离子正极材料晶体结构Fig. 4 Crystal structures of polyanion-based cathode materials.
2.1.2.3 磷酸盐类
Li等56通过氧化LiFePO4制备了FePO4,在1 mol∙L−1Na2SO4中性水溶液电解液中,C/2 倍率下,初始容量约为 110 mAh∙g−1。Minakshi和 Meyrick57首次报道了磷铁钠矿型NaCo1/3Ni1/3Mn1/3PO4(图4c)在水溶液电解液中的活性,发现该材料在有机体系中没有电化学活性,但是在水溶液中却可以实现钠离子的可逆嵌入/脱出58。Deng等59合成了Na7V4(P2O7)4(PO4)/C (图 4d)并首次报道了该材料在水系电解液中的嵌钠性能,发现其具有较高的氧化还原电位(0.961,0.944 V vs SCE),电流密度在 80 mA∙g−1时容量为 51.2 mAh∙g−1,当电流密度增大至1000 mA∙g−1时,容量保持率为72%,但是循环性能有待提高。Vujkovi和 Mentus60通过电化学离子置换法制备了 NaFe0.95V0.05PO4/C,在饱和 NaNO3溶液中,1C (154 mA∙g−1)和 33C 的倍率下,放电容量分别为 127.3,81.9 mAh∙g−1。
2.1.2.4 氟化磷酸盐类
近期研究表明四方相 NaVPO4F (图 4e)在 5 mol∙L−1NaNO3水溶液中具有两个氧化还原峰(0.8,0.2 V vs SCE)61,可作为ARSB正极使用。50 mA∙g−1下,放电容量为 54 mAh∙g−1,但是循环20圈容量便衰减30%。
Kumar等62首次报道了I4/mmm、P42/mnm混合型四方相 Na3V2O2x(PO4)2F3−2x(图 4f–g)作为ARSB正极材料,由于钠离子在水溶液中有限电压范围内的可逆性,容量较低,用 10 mol∙L−1NaClO4水溶液添加 2%碳酸亚乙烯酯(VC)作电解液,电流密度为1C (65 mA∙g−1)时,其容量为46 mAh∙g−1,可稳定循环100圈。该材料具有较高的操作电压(0.71 V vs SCE),适合水系电池的应用。即使在 40C高倍率下,也能保持低倍率(1C)下容量的 40%。随后,该课题组又以 NaTi2(PO4)3-C//Na3V2O2x(PO4)2F3–2x-MWCNT水系全电池体系为例探究了盐浓度和电解液添加剂对电池性能的影响63。
2.1.2.5 焦磷酸盐
Jung等64使用铁基焦磷酸钠 Na2FeP2O7(图4h)作为ARSB正极。与非水电解质相比,由于水溶液电解质中的动力学更快,电池倍率和循环性能得到提高。在−0.654 – +0.576 V (vs SCE)以及2.0–3.8 V (vs Na+/Na)的电压范围内,1C倍率下,在水溶液电解质和非水电解质中,可得到近似的理论容量。在5C下,与水溶液电解质中的1C相比,没有观察到显著的容量降低,并可循环 300圈;然而,在非水电解质中倍率增大到5C后,容量大幅度降低,说明该材料在水溶液电解质中使用具有较大优势。
2.1.3 普鲁士蓝类似物
由于Na+半径相对于Li+较大,因此其在材料中的嵌入和脱出更加困难,此外,Na+在氧化物晶格中相对稳定,当其在材料中嵌入/脱出的时候,会造成材料结构变化。因此采用无氧化物晶格材料是一种好的解决方案。
普鲁士蓝类似物是一种典型的过渡金属铁氰化物,具有三维的开放结构,有利于碱金属离子的传输和储存,可以容纳各种大型阳离子,并且几乎没有晶格畸变。制备的普鲁士蓝类似物薄膜在 Na+等其他碱金属离子基水溶液电解质中均具有电化学活性65–68。此外,在~0.3 V (vs SCE)下与Na发生可逆氧化还原反应,该反应可表示为:
ANiFeⅢ(CN)6⇌ A2NiFeⅡ(CN)6(A = Na 或 K)
在这些研究的基础上,Wessells等69合成了K0.6Ni1.2Fe(CN)6·3.6H2O (NiHCF)块体,并将其用作ARSB正极材料,氧化还原电位为0.59 V (vs SHE),理论容量约为 85 mAh∙g−1。在 1 mol∙L−1NaNO3电解质溶液中,0.83C倍率下,容量约为60 mAh∙g−1,41.7C 时,容量约为 40 mAh∙g–1,在8.3C下循环寿命超过5000次且容量没有衰减。尽管如此,NiHCF电位值较低,在正极应用上优势不足,因此 Wessells等70又对 CuHCF(K0.71Cu[Fe(CN)6]0.72·3.7H2O)进行了类似研究,其表现出较高的电位值(1.0 V vs SHE),41.7C可保持低倍率下容量的34%,8.3C下循环500圈后容量保持率在77%。采用共沉淀法合成的块状CuHCF倍率性能和循环稳定性均得到提升71,72。随后,他们采用共沉淀法合成了CuNiHCF固溶体73,发现CuxNi1−xHCF 氧化还原电位可在 1 mol∙L−1NaNO3水溶液电解液中从0.6 V调至1.0 V。随着Cu含量的增加,电位升高,而且没有明显的容量损失,Cu0.56Ni0.44HCF仍可得到2000圈的循环。Kim等74合成了KCo0.5Cu0.5Fe(CN)6,并用作水系钠离子电池正极,1 A∙g−1电流密度下,可逆容量为 39 mAh∙g−1,并且循环200圈后,可保持该容量的95%,此外还具有较好的倍率性能,10C下,容量为44 mAh∙g−1,100C时,容量可保持在40 mAh∙g−1。虽然这一系列Ni基和Cu基的普鲁士蓝正极材料表现出高倍率性能和循环稳定性,但是由于其本身不含钠(以放电状态存在),故不能与传统的不含 Na的负极材料配对,在用作正极材料时需要先还原至富钠的还原态普鲁士蓝衍生物NaxMFe(CN)6(M = Ni、Co、Fe、Cu 等)。
杨汉西课题组75将含钠的普鲁士蓝Na2NiFe(CN)6用作ARSB正极材料,以Ag/AgCl电极为对电极,在 1 mol∙L−1Na2SO4电解液(pH =7)中,电流密度为 65 mA∙g−1下,Na2NiFe(CN)6容量为 65 mAh∙g−1,10C 下,容量可以维持在~61 mAh∙g−1,5C 下可稳定循环 500圈。制备的Na2CuFe(CN)6显示出大倍率应用前景76,当倍率大至 100C 时,容量为~38 mAh∙g−1,并可在 5C 下稳定循环 500圈。随后,他们又合成了Na2CoFe(CN)677,在 1 mol∙L−1Na2SO4(pH = 7)电解液中表现出两对氧化还原峰,分别代表Co2+/Co3+(+0.40 V vs Ag/AgCl),Fe2+/Fe3+(+0.90 V vs Ag/AgCl)的氧化还原反应,具有较高的初始容量(130 mA∙g−1下放电容量为 128 mAh∙g−1),20C下容量可保持在 61 mAh∙g−1,5C 下循环 800 圈后,容量保持率在 80%。由于普鲁士蓝类似物中存在的水(配位水和沸石水)会阻断Na插入反应活性晶格位点,使得普鲁士蓝及其类似物在水性电解质中的容量利用率较差。为了克服这一问题,该课题组合成了一种低缺陷的普鲁士蓝—FeFe(CN)678(图5),2C 下容量可达 250 mA∙g−1,20C 下,容量仍可保持在102 mAh∙g−1。10C下,循环500圈后,容量保持率为83%。Chen等79将InHCF作为ARSB(M = Li,Na,K)水系可充金属离子电池正极材料进行研究,也得到了较高的功率密度。
最近,向兴德等80在室温下通过沉淀法获得了Ni基普鲁士蓝,通过提高电解液浓度降低了副反应,提高了初始库仑效率、倍率性能和循环稳定性。Paulitsch等81通过沉积法制备了Na2VOx[Fe(CN)6]薄膜作为ARSB正极,排除了阳离子溶剂效应的影响,材料具有较高的半充电电位(ΔE1/2≈ 0.91 V vs Ag/AgCl)以及较好的倍率性能。Lee等82近期合成的 Na0.4(VO)3[Fe(CN)6]·2.12H2O具有多电子反应(V3+↔ V4+↔ V5+,Fe2+↔ Fe3+)特点,在 55 mA∙g−1下,容量为 94.1 mAh∙g−1,当电流密度提高至 3520 mA∙g−1时,容量仍保持在54 mAh∙g−1,在 1760 mA∙g−1的高倍率下循环 1000圈,容量以很低的速度(每圈 0.015%–0.018%)衰减。
尽管这些化学物本身无毒,价格低廉,但制备过程由于CN−的使用,可能会对环境造成影响。此外,合成过程中对水含量的控制十分关键,这将直接影响材料的性能。
2.1.4 有机电极材料

图5 (a) FeFe(CN)6纳米晶体SEM图片,(b) CV曲线,(c) 250 mA∙g−1下充放电曲线以及10C(1C = 125 mA∙g−1)下的循环寿命曲线,(d)倍率性能 78Fig. 5 (a) SEM image of FeFe(CN)6 nanocrystals, (b) CV curves, (c) charge and discharge profiles at a current density of 250 mA∙g−1 and long-term cycling stability 10C rate (1C = 125 mA∙g−1), (d) rate performance 78.
为了克服刚性无机晶格对钠离子的束缚,人们也开始尝试制备有机正极材料。有机电极材料价格低廉,可设计性强,而且一些有机物电极材料可以直接从绿色植物中提取,或进行绿色加工而成,使得有机电极材料的整个循环过程可实现绿色可持续性发展83–85。Koshika等86将自由基聚合物用于水溶液储钠正极,所合成的聚2,2,6,6-四甲基哌啶氧-4-乙烯基醚(PTVE)在 0.1 mol∙L−1NaCl溶液中表现出良好的电化学性能,在0.73 V(vs Ag/AgCl)处呈现出稳定的电压平台,在60C高倍率时的可逆容量达130 mAh∙g−1,循环1000周仍能保持 75%的容量保持率。显然,聚合物储钠材料不仅可以克服无机刚性晶体的束缚,同时大大拓展了材料的选择空间。
2.2 负极材料
2.2.1 活性炭
将活性炭作负极,可以得到混合型水溶液钠离子电容电池,如Whitacre课题组87将Na0.44MnO2作为正极材料,用活性碳做负极,Na2SO4水溶液作为电解液的全电池,在 C/8倍率下电池的比容量为45 mAh∙g−1,4C下循环 1000圈后,容量几乎不变。Liu 等35也将这两种材料组合成全电池,容量为 55 mAh∙g−1(200 F∙g−1),500 mA∙g−1(18C)下循环4000圈后,容量保持率达84%。这类电池虽然能量密度较低,但是结构简单,易于制造,不用选择合适的储钠负极,也是产业化的一种选择方案。
2.2.2 氧化物
MoO3具有层状结构,有利于离子的嵌入和脱出88,并且具有较高的理论容量(1111 mAh∙g−1)89,适合做负极材料,然而其电子导电率较低,结构不稳定90。Deng等91将具有一维纳米结构的Na2V6O16·n H2O用作 ARSB负极材料,氧化还原电位位于−0.4 V (vs SCE),40 mA∙g−1电流密度下的初始容量为123 mAh∙g−1,但是库仑效率较低仅为 45%,并且随着充放电循环进行,容量衰减较快。Vujkovi等92合成的 Na1.2V3O8,具有较快的离子迁移速率和循环稳定性,在NaNO3水溶液中,电位值为−0.67 V (vs SCE),100 mA∙g−1电流密度下,可逆容量为 110 mAh∙g−1。Wang 等93合成了Ti 取代的 Na0.44MnO2(Na0.44[Mn1−xTix]O2)作负极材料,比容量为37 mAh∙g−1,通过Ti取代能够改变充放电平台,减轻电极极化,从而表现出较好的循环稳定性,2C下循环400圈,没有明显的容量衰减。
2.2.3 磷酸盐
NaTi2(PO4)3是水系钠离子电池负极研究方面有潜力的材料,它具有典型的NASICON结构94,由3个PO4四面体和2个TiO6八面体通过角连接组成NaTi2(PO4)3基本单元。一个NaTi2(PO4)3基本单元里存在两种空间位置(A1和A2),其中包括1个A1位点和3个A2位点,钠离子完全占据A1位置,这种开放的三维框架有利于加快钠离子的传输。Delmas等95首先将 NASICON型NaTi2(PO4)3作为非水电解液中的钠嵌入电极。该材料理论容量可达到 133 mAh∙g−1,在 1 mol∙L−1Na2SO4溶液中,NaTi2(PO4)3在−0.82 V (vs Ag/AgCl)处表现出一对十分可逆且对称的氧化还原峰,并且平台长而平坦,对应于钠离子的可逆嵌脱反应96。这一反应电势区接近但略高于水的析氢电位,可确保正常的嵌钠反应并且没有析氢副反应的发生,有利于提高电池的工作电压,从而获得较大的电压输出。但是,该材料电子导电率较低,并且在水溶液中受到pH影响96–98,因此需要通过碳包覆等方法进行改进。Whitacre等99通过微波辅助法合成了NaTi2(PO4)3(图6a),优化后的 NaTi2(PO4)3在 15.7 mA∙g−1电流密度下具有85 mAh∙g−1,并且可循环20圈。随后他们又合成了含碳纳米管(CNTs)和石墨(G)包覆的NaTi2(PO4)3(图 6b)100,0.1C下比容量达到 130 mAh∙g−1。
与Na0.44MnO2组成全电池,2C下可获得理论容量的56%。NaTi2(PO4)3/G + 碳纳米管(CNTs)表现出较好的循环性能,1C下循环100圈后容量保持率在86%。Pang等94合成了NaTi2(PO4)3-石墨烯纳米复合物(图6c),提高了高倍率下的比容量,2C和 10C下可分别获得比容量为 110及 65 mAh∙g−1,2C下循环 100圈容量保持率为 95%。该课题组也将NaTi2(PO4)3与多壁碳纳米管(MWNTs)复合(图 6d)101,提高了循环性能与倍率性能。钱逸泰组102通过包覆石墨烯改性了NaTi2(PO4)3(图6e),20C倍率下,比容量 63.5 mAh∙g−1,循环 2000次后容量保持率为 71%。Zhao 等103通过结构和碳包覆双重改性制备了蛙卵状分层多孔NaTi2(PO4)3碳阵列(图 6f),提高了材料的电化学性能,90C下容量为 66 mAh∙g−1,并且 20C倍率下循环2000圈后,容量保持率在89%。Hung等104通过水热法合成了NaTi2(PO4)3纳米粒子(图6g),0.2C下容量为 121 mAh∙g−1,1C下循环 300 圈后容量保持率在75%。He等105通过改进的Pechini法制备了NaTi2(PO4)3,并通过热解法包覆碳(图6h),表现出较高倍率性能和循环稳定性。通过碳的包覆改性,不仅提高了NaTi2(PO4)3电子电导率,也可以减缓材料表面与电解液的接触,防止副反应的发生,从而提高材料的嵌脱离子性能,在全电池应用上也具有较好性能。

图6 不同NaTi2(PO4)3 SEM或TEM图像Fig. 6 SEM or TEM images of different NaTi2(PO4)3.
Na3Ti2(PO4)3也被用作ARSB负极材料56,由于该材料预嵌钠,可以与不含钠的正极材料进行匹配。但该使用条件苛刻,必须在无氧条件下进行。此外,NASICON结构的NaV3(PO4)3,Na2VTi(PO4)3,Na3MnTi(PO4)3也可以用做负极。最近,Ke等106通过静电纺丝法合成了平行排列的NaV3(PO4)3@C纳米纤维,得到了较好的倍率性能,在 1C和 25C倍率下可分别得到 118,63 mAh∙g−1比容量,但是由于该材料的电位较高,因此得到较高的输出电压具有挑战性。
Minakshi和Ralph107通过sol-gel法合成了NaCo1/3Ni1/3Mn1/3PO4,用MnO2做正极,使用含1 mol∙L−1ZnSO4的 7 mol∙L−1NaOH作电解液的全电池表明可以进行钠离子的可逆脱嵌,容量为 250 mAh∙g−1,但是电压较低。
2.2.4 普鲁士蓝类似物
Pasta等108报道了锰基普鲁士蓝(K0.11Mn[Mn(CN)6]0.83·3.64H2O)作为ARSB负极材料,氧化还原电位为0.052 V (vs SHE),比容量为57 mAh∙g−1,从 1C到 5C再到 10C,容量几乎不变。
2.2.5 有机物
同样,为了突破无机材料的限制,近年来,人们也开展了有机储钠负极的研究。Qin等61将1,4,5,8-萘四羧酸二酐(NTCDA)衍生物——聚酰亚胺作为ARSB负极材料,其结构中共轭羰基基团可以发生可逆的氧化还原反应,从而实现电荷储存。在5 mol∙L−1NaNO3电解液中的平均充电电位、放电电位分别为−0.50 和−0.39 V(vs SCE),50 mA∙g−1下,充放电容量分别为184和165 mAh∙g−1,循环20圈后,容量衰减 17%。夏永姚课题组7通过NTCDA制备了聚酰亚胺(PNTCDA),将其作为负极使用,与I−/I3−正极构成全电池,电荷通过钠离子、碘离子在正负极间迁移而传递,1 A∙g−1(5.5C)下,放电容量为 140 mAh∙g−1,当电流密度加大到 40 A∙g–1(220C)时容量仍可达 59 mAh∙g–1,并且循环50000圈后,容量保持率仍有70%。该体系将电池的高能量密度和电容器的高功率密度有效结合起来,表现出高功率性能。Choi等109设计合成了蒽醌结构聚合物[poly(2-vinylanthraquinone),PVAQ],证实了在蒽醌基团两电子还原过程中通过两个钠离子嵌入聚合物链段以维持电荷平衡。这种聚合物在pH = 14的NaCl水溶液中表现出平稳的电压平台。以5 A∙g−1电流密度充放电,可逆容量高达217 mAh∙g−1,循环 300次后容量保持率为 91%,这一性能可以基本达到应用要求。Kim等74将萘二甲酸二钠(H) SNDI用作ARSB负极,1 mol∙L−1Na2SO4(pH = 7)水溶液中,平均氧化还原电位在−0.04 V (vs SHE),6C下可逆容量为 62 mAh∙g−1,24C时,可逆容量仍有 40 mAh∙g–1。6C下循环 500圈后,容量保持率有 74%。Liang等110使用醌基芘-4,5,9,10-四酮(PPTO)做负极,与Na3V2(PO4)3构成全电池,1C时容量为 201 mAh∙g−1,能量密度达30 Wh∙kg−1,循环80圈后,容量保持率在79%。
2.3 全电池
在上述ARSB的电极材料(正极、负极)基础上,人们也开始了水系钠离子全电池体系的研究。
正极材料主要集中在金属氧化物、聚阴离子化合物和普鲁士蓝上,而锰氧化物与NaTi2(PO4)3的组合被广泛研究。如Liu等111将包含碱金属的层状纳米结构δ-MnO2(A-δ-MnO2, A = K+, Na+)(K : Na : Mn的原子比为0.15 : 0.26 : 1)作为ARSB正极材料,NaTi2(PO4)3作为负极材料的全电池(图7a),在电流密度为150 mA∙g−1时,比容量为74.6 mAh∙g−1,在 600 mA∙g−1的高电流密度下,容量保持率在~62%,并且 200 mAh∙g−1下,循环 200 圈后没有明显衰减。Li等人112构筑了Na0.44MnO2/NaTi2(PO4)3-C水系钠离子电池,如图7b所示,倍率性能非常优异(3C–270C),在5C–50C间循环700周,容量保持率为60%。而且可以稳定循环1500周以上,以整个电池质量计算,能量密度达到65 Wh∙L−1。Zhang等113通过水热辅助sol-gel法合成了Na3V2(PO4)3,用NaTi2(PO4)3做负极,Na2SO4作电解液,可构成1.2 V的全电池(图7c),具有较好的倍率性能。在 10 mA∙g−1的电流密度下,容量为58 mAh∙g−1。5145 W∙kg−1功率下,电池容量密度达到 29 Wh∙kg−1。崔屹课题组108将CuHCF和MnHCF作为正负极(图 7d–e),在 10 mol∙L−1NaClO4水溶液电解液中,可得到0.95 V的开路电压,10C下循环1000圈后几乎没有容量衰减。
负极方面,采用高表面的活性炭材料作为负极,嵌钠化合物作为正极,可构成混合型水系钠离子电容电池。如Whitacre等114先合成尖晶石型LiMn2O4,经过化学脱锂制备了λ-MnO2。分别使用Na0.44MnO2和λ-MnO2作为正极,活性炭作为负极的水系钠离子二次电池(图7f)。发现水溶液中尖晶 石 型 λ-MnO2的 比 容 量 (~80 mAh∙g–1) 为Na0.44MnO2的2倍以上,比能量增加3倍。NaTi2(PO4)3由于具有较合适的输出电压(1.2 V),较长的循环寿命(> 1000圈),以及较好的适应性,成为ARSB中比较具有吸引力的材料,很多正极材料将其作为匹配负极(表2)。此外,金属氧化物具有层状结构,有利于离子的嵌入和脱出88,并且具有较高的理论容量(1111 mAh∙g−1)89。如Liu等116合成了聚吡咯包覆的MoO3(PPy@MoO3),与Na0.35MnO2构成的全电池具有较好的倍率性能和循环稳定性,2.6 kW∙kg−1功率密度下,能量密度可保持 18 Wh∙kg−1。但这类氧化物成本高,循环寿命短,倍率性能不足,难以进行工业化应用。其他负极材料如有机物也得到研究,如Kim等74用SNDI作负极,KCo0.5Cu0.5Fe(CN)6作正极,可得到1.1 V的开路电压(图7g)。

图7 全电池的充放电曲线、CV曲线和实物图Fig. 7 Charge/discharge profiles, CV curves and digital photos of full batteries.
整体看来,目前所研究的全电池体系的能量密度一般在 50 Wh∙kg–1以下。毫无疑问,未来能源经济必须立足于绿色、廉价、可持续的能源供应,大型可充电电池也因此成为用来存储可持续能源的储能设备,但它与用在移动电子设备上的小型电池的要求不同,其必须具备高安全、低成本和长寿命的特点。与有机电解液体系相比,水系钠离子电池的能量密度虽然比较小,但它具有高安全性和低成本的优点,并且在大多数固定式储能场合,能量密度并非首要考虑的因素,成本与安全性通常是更为关心的指标,因此研制适合于规模储能的水系钠离子电池体系才是未来发展的重点。
为了系统比较主要电极材料的电化学性质,表 2列出了典型水系钠离子电池全电池体系,主要介绍了其构造和电化学性能,对加速钠离子电池的实用化进程具有一定参考价值。在实际应用上,有机钠离子电池方面,国内外知名公司如日本住友电气工业公司、英国Faradion公司、Sharp Laboratories of America (SLA)以及国内中科院物理所和上海交通大学与相关公司已经开展了有机钠离子电池商业化的前期工作。而美国Aquion Energy公司、国内恩力能源科技更将水系钠离子电池推向了市场。其中Jay F. Whitacre创办的美国Aquion Energy公司是全球第一家批量生产水系钠离子电池的公司,该公司采用水性电解液Na2SO4和储量十分丰富的钠和锰(Na0.44MnO2)开发了水系钠离子电池。这种电池成本低廉(300美元/kWh),不到锂离子电池使用成本的三分之一。第三方测试表明,Aquion Energy的电池可以实现持续5000次以上的充放电循环,且效率均在85%以上。

表2 一些典型的ARSB全电池的电化学性能Table 2 Electrochemical Properties of Representative Full Cell Configurations for ARSB.

continued Table 2
2015年4月16日,我国第一条拥有自主知识产权的水系离子电池生产线在江苏南通—恩力能源科技(南通)有限公司顺利投产。这也标志着我国拥有了具有自主知识产权、居世界领先水平的水系离子储能产品和储能系统。恩力第一代20 MWh生产线生产出的水系离子电池组成储能系统,可保证每个充电放电循环的成本低于 0.19美元/度电,相比锂电池系统和铅酸电池系统具有优势,并且低于发达国家电网峰谷电价差额,在电网用电端使用具有经济性(谷电价段存电储能,峰电价段用电)。这种电池循环寿命目前已经达到 3000次以上;接近100%深度放电;可以确保在10年内正常使用。
3 挑战与展望
水系钠离子电池资源储备丰富,环境友好,安全可靠,尽管钠原子质量较锂重并且电位稍高于锂(0.33 V vs Li),但是其比能量成本低的优势足以抵消这些缺点,如制备含钠电极材料以及电解液都比锂相对便宜,并且可以用便宜而轻质的Al集流体代替Cu集流体而实现低成本和高的比能量,因此可以替代常规可再充电池进行大规模应用。然而,在投入实际应用之前,还存在一些挑战,如:
(1) 由于水的电化学窗口狭窄,水系钠离子电池电极材料的选择范围受限,需要进一步的探索。
在正极材料中,Na0.44MnO2由于其高的化学稳定性和低成本而被广泛研究。在不进行包覆的情况下可以稳定循环4000次。然而,对于锰基氧化物来说,由于可利用的钠离子数量有限,锰基氧化物比容量较低,Na0.44MnO2一般在 45 mAh∙g−1左右,总体能量密度偏低。
为此,人们寻找替代材料以克服这一问题,如MnO2多晶型物等。其中,λ-MnO2材料具有~80 mAh∙g−1的比容量,循环稳定性高达5000次循环。然而,λ-MnO2通常由LiMn2O4电化学脱锂制备,工艺繁琐。非氧化物晶格材料—普鲁士蓝类似物具有开放的框架结构,使得Na+可以在其中快速扩散,得到高容量和良好的循环性,具有较大优势,例 如 , MnII-N≡C-MnIII在 MnII-N≡C-MnIII//CuII-N≡C-FeIII全电池中可稳定循环1000次且没有明显的容量衰减,但是需要高饱和电解液。普鲁士蓝类似物通常需要在pH < 2的电解液中工作,影响电池容量,并且首圈库仑效率较低,在提高性能方面需要进一步的研究。
聚阴离子化合物基电极材料由于结构的多样性使其具有较宽的正负极电压选择范围,NASICON型NaTi2(PO4)3是ARSB少数负极材料中最具代表性的。理论容量为~133 mAh∙g−1,电位平台约−0.6 V (vs SHE)。但反应过程中也会发生H2析出反应,使得循环稳定性差。此外,NaTi2(PO4)3电子导电性较低,需要经过碳包覆等改性以获得稳定的电化学性能,会降低材料的能量密度。因此,可以设计微纳结构及薄层碳包覆结构的聚阴离子基材料。制备新型聚阴离子材料也是一个有意义的研究方向。
与无机化合物相比,有机类电极极材料资源丰富,种类繁多,氧化还原电位可调节的范围较宽,可发生多电子反应,可设计强,且具有结构上的韧性,灵活性更高,是未来重要的发展方向。
(2) 多种副反应在水系电解液中会与电极材料发生反应降低电池可逆性。如水分解过程中的析氢、析氧反应;嵌钠态的负极材料化学反应活性高,易与水发生反应;电极材料与H2O和电解液中残余 O2的反应;电极材料的溶解;质子 H+在电极材料中与离子共嵌入造成变价金属离子的歧化和溶解,形成电化学惰性产物;电极材料晶体缺陷、溶解和充放电过程中引起的晶体结构变化,集流体的氧化腐蚀等。因此应提出一些电极保护措施。如Whitacre等研究表明高浓度电解质可以提高电池倍率性能。王春生等提出了一种“Water in Salt”的新型电解液,通过这种超饱和电解质,在负极上形成致密的固体电解质界面(SEI),水的电化学活性降低,从而扩大电化学稳定性窗口,提高能量密度。然而,高浓度电解质会引起电极材料等的腐蚀问题,特别是在极端的化学势下,这也是一个挑战。
(3) 电极本身催化效应,钠盐的种类,添加剂,pH值,离子导电性均会影响 H2、O2的析出和电极材料的稳定性,进而影响反应效率和循环稳定性,特别是纳米结构材料大的比表面积更加速副反应发生,因此需要对材料进行表面包覆等改进或加入添加剂以稳定电极材料。此外,也需要进一步研究不同集流体在电解液中的效应以延长电池循环寿命。
目前水系钠离子电池面临的主要问题是亟需提高电极材料在水溶液中的循环稳定性并以此提高电池的循环寿命。今后对水系钠离子电池的研究应致力以下方面:(1) 水溶液中电极材料容量衰减的机理;(2) 正负极材料的选取、匹配和配比的优化以及新型水溶液中可嵌/脱钠离子材料的开发;(3) 电极材料的表面修饰以及掺杂改性,以提高其在水溶液中的稳定性,提高电极过电位;(4)选择与电极材料相匹配的电解液(合适的pH范围、盐的种类和浓度)和电解液添加剂的研究;(5) 选择合适的集流体(优良导电性、较高机械强度、较高析氢、析氧过电位和良好的耐腐蚀性能)。
最近的研究提出,具有“开放框架”或有机材料的主体材料能够通过化学键的重新排列可逆地吸收和释放其柔性结构中的客体离子,从而提高水系钠离子电池系的稳定性。此外还可以使用上述若干体系设计来解决当前的挑战,包括采用多价离子,氧气电极和混合电解液等。
据立木信息咨询发布的《中国水系钠离子电池市场预测及战略研究报告(2017版)》显示:水系钠离子电池是非常适合用于储能领域的一种电池,其 70% DOD深充放循环使用寿命超过 3000次,不使用重金属元素,且由于钠资源地球储量极其丰富,未来批量生产售价可低至150美元/(kW h),表明水系钠离子电池为高安全,低成本,大规模的可持续能源利用提供了解决方法。随着越来越多的电极材料的发现和评估,相信ARSB的综合性能将不断提高。预计在不久的将来,ARSB将在大规模固定储能中发挥重要作用,推动智能电网和大规模可再生能源并网的清洁能源的使用和发展。
(1) Yang, Z.; Zhang, J.; Kintner-Meyer, M. C. W.; Lu, X.; Choi, D.;Lemmon, J. P.; Liu, J. Chem. Rev. 2011, 111, 3577.doi: 10.1021/cr100290v
(2) Dunn, B.; Kamath, H.; Tarascon, J. M. Science 2011, 334, 928.doi: 10.1126/science.1212741
(3) Wen, Y.; He, K.; Zhu, Y. J.; Han, F. D.; Xu, Y. H.; Matsuda, I.; Ishii,Y.; Cumings, J.; Wang, C.S. Nat. Commun. 2014, 5, 4033.doi: 10.1038/ncomms5033
(4) Slater, M. D.; Kim, D.; Lee, E.; Johnson, C. S. Adv. Funct. Mater.2013, 23, 947. doi: 10.1002/adfm.201200691
(5) Kundu, D.; Talaie, E.; Duffort, V.; Nazar, L. F. Angew. Chem. Int. Ed.2015, 54, 3431. doi: 10.1002/anie.201410376
(6) Fang, Y. J.; Chen, Z. X.; Ai, X. P.; Yang, H. X.; Cao, Y. L. Acta Phys. -Chim. Sin. 2017, 33, 211. [方永进, 陈重学, 艾新平, 杨汉西, 曹余良. 物理化学学报, 2017, 33, 211.]doi: 10.3866/PKU.WHXB201610111
(7) Dong, X. L.; Chen, L.; Liu, J. Y.; Haller, S.; Wang, Y. G.; Xia, Y. Y.Sci. Adv. 2016, 2, e1501038. doi: 10.1126/sciadv.1501038
(8) Yang, H. X.; Qian, J. F. J. Inorg. Mater. 2013, 28, 1165. [杨汉西,钱江锋. 无机材料学报, 2013, 28, 1165.]doi: 10.3724/SP.J.1077.2013.13388
(9) Zhang, N.; Liu, Y. C.; Chen, C. C.; Tao, Z. L.; Chen, J.; Chin.J. Inorg. Chem. 2015, 31, 1739. [张宁, 刘永畅, 陈程成, 陶占良,陈军. 无机化学学报, 2015, 31, 1739.] doi: 10.11862/cjic.2015.258
(10) Tang, W.; Zhu, Y.; Hou, Y.; Liu, L.; Wu, Y.; Loh, K. P.; Zhang, H.;Zhu, K. Energy Enviorn. Sci. 2013, 6, 2093.doi: 10.1039/C3EE24249H
(11) Li, W.; Dahn, J. R.; Wainwright, D. S. Science 1994, 264, 1115.doi: 10. 1126/science.264.5162.1115
(12) Cao, Y.; Wang, Y. G.; Wang, Q.; Zhang, Z. Y.; Chen, Y.; Xia, Y. Y.;Dai, X. Energy Storage Sci. Technol. 2016, 5, 317. [蓸翊, 王永刚,张青, 张兆勇, 车勇, 夏永姚, 戴翔. 储能科学与技术, 2016, 5,317.] doi: 10.3969/j.issn.2095-4239.2016.03.008
(13) Kim, H.; Hong, J.; Park, K. Y.; Kim, H.; Kim, S. W.; Kang, K. Chem.Rev. 2014, 114, 11788. doi: 10.1021/cr500232y
(14) Lu, Y.; Goodenough, J. B.; Kim, Y. J. Am. Chem. Soc. 2011, 133,5756. doi:10.1021/ja201118f
(15) Luo, J. Y.; Cui, W. J.; He, P.; Xia, Y. Y. Nat. Chem. 2010, 2, 760.doi: 10.1038/nchem.763
(16) Ghodbane, O.; Pascal, J. L.; Favie, F. ACS Appl. Mater. Interfaces 2009, 1, 1130. doi: 10.1021/am900094e
(17) Hill, L. I.; Verbaere, A.; Guyomard, D. J. Power Sources 2003, 119–121, 226. doi: 10.1016/S0378-7753(03)00238-6
(18) Cao, J.; Mao, Q. H.; Shi, L.; Qian, Y. T. J. Mater. Chem. 2011, 21,16210. doi: 10.1039/C1JM10862J
(19) Kitchaev, D. A.; Dacek, S. T.; Sun, W. H.; Ceder, G. J. Am. Chem.Soc. 2017, 139, 2672. doi: 10.1021/jacs.6b11301
(20) Tarascon, J. M.; Guyomard, D. G.; Wilkens, B.; Mc Kinnon, W. R.;Barboux, P. Solid State Ionics 1992, 57, 113.doi: 10.1016/0167-2738(92) 90072-W
(21) Kanoh, H.; Tang, W.; Makita, Y.; Ooi, K. Langmuir 1997, 13, 6845.doi: 10.1021/la970767d
(22) Athouel, L.; Moser, F.; Dugas, R.; Crosnier, O.; Belanger, D.;Brousse, T. J. Phys. Chem. C 2008, 112, 7270.doi: 10.1021/jp0773029
(23) Athouel, L.; Moser, F.; Dugas, R.; Crosnier, O.; Belanger, D.;Brousse, T. ECS Trans. 2008, 16, 119. doi: 10.1149/1.2985634
(24) Shao, J.; Li, X. Y.; Qu, Q. T.; Wu, Y. P. J. Power Sources 2013, 223,56. doi: 10.1016/j.jpowsour.2012.09.046
(25) Komaba, S.; Ogata, A.; Tsuchikawa, T. Electrochem. Commun. 2008,10, 1435. doi: 10.1016/j.elecom.2008.07.025
(26) Minakshi, M. Mater. Sci. Eng. B 2012, 177, 1788.doi: 10.1016/j.mseb.2012.09.003
(27) Qu, Q. T.; Liu, L. L.; Wu, Y. P.; Holze, R. Electrochim. Acta 2013,96, 8. doi: 10.1016/j.electacta.2013.02.078
(28) Sun, X. Structures and Electrochemical Performances of Transition Metal Oxides NaMO2as Electrode Materials for Sodium-Ion Batteries. Ph. D. Dissertation, University of Science and Technology of China, Anhui, 2016. [孙信. 过渡金属氧化物NaxMO2结构调控和储钠性能的研究[D]. 合肥: 中国科学技术大学, 2016.]
(29) Su, D. W.; Wang, C. Y.; Ahn, H. J.; Wang, G. X. Chem. Eur. J. 2013.19, 10884. doi: 10.1002/chem.201301563
(30) Liu, Y. C.; Chen, C. C.; Zhang, N.; Wang, L. B.; Xiang, X. D.; Chen,J. J. Electrochem. 2016, 22, 437. [刘永畅, 陈程成, 张宁, 王刘彬,向兴德, 陈军. 电化学, 2016, 22, 437.]doi: 10.13208/j.electrochem.160548
(31) Sauvage, F.; Baudrin, E.; Tarascon, J. M. Sens. Actuators, B 2007,120, 638. doi: 10.1016/j.snb.2006.03.024
(32) Parant, J. P.; Olazcuaga, R.; Devalette, M.; Fouassier, C.;Hagenmuller, P. J. Solid State Chem. 1971, 3, 1.doi: 10.1016/0022-4596(71)90001-6
(33) Kim, H.; Kim, D. J.; Seo, D. H.; Yeom, M. S.; Kang, K.; Kim, D. K.;Jung, Y. Chem. Mater. 2012, 24, 1205. doi: 10.1021/cm300065y
(34) Kim, D. J.; Ponraj, R.; Kannan, A. G.; Lee, H. W.; Fathi, R.; Ruffo,R.; Mari, C. M.; Kim, D. K. J. Power Sources 2013, 244, 758.doi: 10.1016/j.jpowsour.2013.02.090
(35) Liu, X.; Zhang, N.; Ni, J.; Gao, L. J. Solid State Electrochem. 2013,17, 1939. doi: 10.1007/s10008-013-2044-0
(36) Dai, K.; Mao, J.; Song, X.; Battaglia, V.; Liu, G. J. Power Sources 2015, 285, 161. doi: 10.1016/j.jpowsour.2015.03.087
(37) Zhang, B. H.; Liu, Y.; Chang, Z.; Yang, Y. Q.; Wen, Z. B.; Wu, Y. P.;Holze, R. J. Power Sources 2014, 253, 98.doi: 10.1016/j.jpowsour.2013.12.011
(38) Tevar, A. D.; Whitacre, J. F. J. Electrochem. Soc. 2010, 157, A870.doi: 10.1149/1.3428667
(39) Zhang, X. Q.; Hou, A. G.; Li, A. N.; Liang, A. W.; Zhu, Y. C.; Qian,Y. T. J. Mater. Chem. A 2016, 4, 856. doi: 10.1039/C5TA08857G
(40) Yu, F.; Zhang, S. M.; Fang, C.; Liu, Y.; He, S. Y.; Xia, J.; Yang, J. H.;Zhang, N. Ceram. Int. 2017, 43, 9960.doi: 10.1016/j.ceramint.2017.05.007
(41) Liu, Y.; Qiao, Y.; Zhang, W.; Xu, H.; Li, Z.; Shen, Y.; Yuan, L.; Hu,X.; Dai, X.; Huang, Y. H. Nano Energy 2014, 5, 97.doi: 10.1016/j.nanoen.2014.02.010
(42) Liu, Y.; Qiao, Y.; Lou, X. F.; Zhang, X. H.; Huang, Y. H. ACS Appl.Mater. Inter. 2016, 8, 14564. doi: 10.1021/acsami.6b03089
(43) Wang, Y. S.; Mu, L. Q.; Liu, J.; Yang, Z. Z.; Yu, X. Q.; Gu, L.; Hu, Y.S.; Li, H.; Yang, X. Q.; Chen, L. Q.; et al. Adv. Energy Mater. 2015,5, 1501005. doi: 10.1002/aenm.201501005
(44) Jung, Y. H.; Hong, S. T.; Kim, D. K. J. Electrochem. Soc. 2013, 160,A897. doi: 10.1149/2.113306jes
(45) Andersson, A. S.; Kalska, B.; Haggstrom, L.; Thomas, J. O. Solid State Ionics 2000, 130, 41. doi: 10.1016/S0167-2738(00)00311-8
(46) Padhi, A. K.; Nanjundaswamy, K. S.; Masquelier, C.; Goodenough, J.B. J. Electrochem. Soc. 1997, 144, 2581. doi: 10.1149/1.1837868
(47) Tarascon, J. M.; Armand, M. Nature 2001, 414, 359.doi: 10.1038/35104644
(48) Song, W. X.; Hou, H. S.; Ji, X. B. Acta Phys. -Chim. Sin. 2017, 33,103. [宋维鑫, 侯红帅, 纪效波. 物理化学学报, 2017, 33, 103.]doi: 10.3866/PKU.WHXB201608303
(49) Song, W. W.; Ji, X. B.; Zhu, Y.; Zhu, H. J.; Li, F. Q.; Chen, J.; Lu, F.;Yao, Y. P.; Banks, C. E. ChemElectroChem 2014, 1, 871.doi: 10.1002/celc.201300248
(50) Mason, C. M.; Lange, F. ECS Electrochem. Lett. 2015, 4, A79.doi: 10.1149/2.0011508eel
(51) Fernandez-Ropero, A. J.; Saurel, D.; Acebedo, B.; Rojo, T.;Casas-Cabanas, M. J. Power Sources 2015, 291, 40.doi: 10.1016/j.jpowsour.2015.05.006
(52) Vujkovic, M.; Mentus, S. J. Power Sources 2014, 247, 184.doi: 10.1016/j.jpowsour.2013.08.062
(53) Levi, M. D.; Sigalov, S.; Salitra, G.; Elazari, R.; Aurbach, D.;Daikhin, L.; Presser, V. J. Phys. Chem. C 2013, 117, 1247.doi: 10.1021/jp3117819
(54) Zhao, Z. W.; Si, X. F.; Liang, X. X.; Liu, X. H.; He, L. H. Trans.Nonferrous Met. Soc. China 2013, 23, 1157.doi: 10.1016/S1003-6326(13)62578-9
(55) Moreau, P.; Guyomard, D.; Gaubicher, J.; Boucher, F. Chem. Mater.2010, 22, 4126. doi: 10.1021/cm101377h
(56) Li, Z.; Ravnsbaek, D. B.; Xiang, K. B.; Chiang, Y. M. Electrochem.Commun. 2014, 44, 12. doi:10.1016/j.elecom.2014.04.003
(57) Minakshi, M.; Meyrick, D. J. Alloys Compd. 2013, 555, 10.doi: 10.1016/j.jallcom.2012.11.203
(58) Minakshi, M.; Meyrick, D.; Appadoo, D. Energ. Fuel. 2013, 27,3516. doi: 10.1021/ef400333s
(59) Deng, C.; Zhang, S.; Wu, Y. X. Nanoscale 2015, 7, 487.doi: 10.1039/C4NR05175K
(60) Vujkovic, M.; Mentus, S. J. Power Sources 2016, 325, 185.doi: 10.1016/j.jpowsour.2016.06.031
(61) Qin, H.; Song, Z. P.; Zhan, H.; Zhou, Y. H. J. Power Sources 2014,249, 367. doi:10.1016/j.jpowsour.2013.10.091
(62) Kumar, P. R.; Jung, Y. H.; Lim, C. H.; Kim, D. K. J. Mater. Chem. A 2015, 3, 6271. doi: 10.1039/C5TA00980D
(63) Kumar, P. R.; Jung, Y. H.; Moorthy, B.; Kim, D. K. J. Electrochem.Soc. 2016, 163, A1484. doi: 10.1149/2.0031608jes
(64) Jung, Y. H.; Lim, C. H.; Kim, J. H.; Kim, D. K. RSC Adv. 2014, 4,9799. doi: 10.1039/C3RA47560C
(65) Bocarsly, A. B.; Sinha, S. J. Electroanal. Chem. 1982, 137, 157.doi: 10.1016/0022-0728(82)85075-4
(66) Bocarsly, A. B.; Sinha, S. J. Electroanal. Chem. 1982, 140, 167.doi: 10.1016/0368-1874(82)85310-0
(67) Itaya, K.; Uchida, I.; Neff, V. D. Acc. Chem. Res. 1986, 19, 162.doi: 10.1021/ar00126a001.
(68) Kalwellis-Mohn, S.; Grabner, E. W. Electrochim. Acta 1989, 34,1265. doi:10.1016/0013-4686(89)87169-5
(69) Wessells, C. D.; Peddada, S. V.; Huggins, R. A.; Cui, Y. Nano Lett.2011, 11, 5421. doi: 10.1021/nl203193q
(70) Wessells, C. D.; Peddada, S. V.; McDowell, M. T.; Huggins, R. A.;Cui, Y. J. Electrochem. Soc. 2012, 159, A98.doi: 10.1149/2.060202jes
(71) Wessells, C. D.; Huggins, R. A.; Cui, Y. Nat. Commun. 2011, 2, 550.doi: 10.1038/ncomms1563
(72) Pasta, M.; Wessells, C. D.; Huggins, R. A.; Cui, Y. Nat. Commun.2012, 3, 1149. doi: 10.1038/ncomms2139
(73) Wessells, C. D.; McDowell, M. T.; Peddada, S. V.; Pasta, M.;Huggins, R. A.; Cui, Y. ACS Nano 2012, 6, 1688.doi: 10.1021/nn204666v
(74) Kim, D. J.; Jung, Y. H.; Bharathi, K. K.; Je, S. H.; Kim, D. K.;Coskun, A.; Choi, J. K. Energy Mater. 2014, 4, 1400133.doi: 10.1002/aenm.201400133
(75) Wu, X. Y.; Cao, Y. L.; Ai, X. P.; Qian, J. F.; Yang, H. X. Electrochem.Commun. 2013, 31, 145. doi: 10.1016/j.elecom.2013.03.013
(76) Wu, X. Y.; Sun, M. Y.; Shen, Y. F.; Qian, J. F.; Cao, Y. L.; Ai, X. P.;Yang, H. X. ChemSusChem 2014, 7, 407.doi: 10.1002/cssc.201301036
(77) Wu, X. Y.; Sun, M. Y.; Guo, S. M.; Qian, J. F.; Liu, Y.; Cao, Y. L.; Ai,X. P.; Yang, H. X. ChemNanoMat 2015, 1, 188.doi: 10.1002/cnma.201500021
(78) Wu, X. Y.; Luo, Y.; Sun, M. Y.; Qian, J. F.; Cao, Y. L.; Ai, X. P.;Yang, H. X. Nano Energy 2015, 13, 117.doi: 10.1016/j.nanoen.2015.02.006
(79) Chen, L.; Shao, H. Z.; Zhou, X. F.; Liu, G. Q.; Jiang, J.; Liu, Z. P.Nat. Commun. 2016, 7, 11982 .doi: 10.1038/ncomms11982
(80) Li, W. F.; Zhang, F.; Xiang, X. D.; Zhang, X. C. ChemElectroChem 2017, 4, 2870. doi: 10.1002/celc.201700776
(81) Paulitsch, B.; Yun, J.; Bandarenka, A. S. ACS Appl. Mater.Interfaces 2017, 9, 8107. doi: 10.1021/acsami.6b15666
(82) Lee, J. H.; Ali, G.; Kim, D. H.; Chung, K. Y. Adv. Energy Mater.2017, 7, 1601491. doi: 10.1002/aenm.201601491
(83) Zhu, Z. Q.; Li, H.; Liang, J.; Tao, Z. L.; Chen, J. Chem. Commun.2015, 51, 1446. doi: 10.1039/C4CC08220F
(84) Guo, C. Y.; Zhang, K.; Zhao, Q.; Pei, L. K.; Chen, J. Chem.Commun. 2015, 51, 10244. doi: 10.1039/C5CC02251G
(85) Wang, S. W.; Wang, L. J.; Zhang, K.; Zhu, Z. Q.; Tao, Z. L.; Chen,J. Nano Lett. 2013, 13, 4404. doi: 10.1021/nl402239p
(86) Koshika, K.; Sano, N.; Oyaizu, K.; Nishide, H. Chem. Commun.2009, 7, 836. doi:10.1039/b818087c
(87) Whitacre, J. F.; Tevar, A.; Sharma, S. Electrochem. Commun. 2010,12, 463. doi: 10.1016/j.elecom.2010.01.020
(88) Mai, L. Q.; Hu, B.; Chen, W.; Qi, Y. Y.; Lao, C. S.; Yang, R. S.; Dai,Y.; Wang, Z. L. Adv. Mater. 2007, 19, 3712.doi: 10.1002/adma.200700883
(89) Xia, X. F.; Hao, Q. L.; Lei, W.; Wang, W. J.; Wang, H. L.; Wang, X.J. Mater. Chem. 2012, 22, 8314. doi: 10.1039/C2JM16216D
(90) Zhou, L.; Yang, L. C.; Yuan, P.; Zou, J.; Wu, Y. P.; Yu, C. Z. J. Phys.Chem. C 2010, 114, 21868. doi: 10.1021/jp108778v
(91) Deng, C.; Zhang, S.; Dong, Z.; Shang, Y. Nano Energy 2014, 4, 49.doi: 10.1016/j.nanoen.2013.12.014
(92) Vujkovic, M.; Paunkovic, B. S.; Simatovic, I. S.; Mitric, M.;Sequeira, C. A. C.; Mentus. S. Electrochim. Acta 2014, 147, 167.doi: 10.1016/j.electacta.2014.08.137
(93) Wang, Y. S.; Liu, J.; Lee, B.; Qiao, R.; Yang, Z. Z.; Xu, S. Y.; Yu, X.Q.; Gu, L.; Hu, Y. S.; Yang, W. L.; et al. Nat. Commun. 2015, 6,6401. doi: 10.1038/ncomms7401
(94) Pang, G.; Yuan, C. A.; Nie, P.; Ding, B.; Zhu, J. J.; Zhang, X. G.Nanoscale 2014, 6, 6328. doi: 10.1039/C3NR06730K
(95) Delmas, C.; Cherkaoui, F.; Nadiri, A.; Hagenmuller, P. Mater. Res.Bull. 1987, 22, 631. doi: 10.1016/0025-5408(87)90112-7
(96) Park, S.; II; Gocheva, I.; Okada, S.; Yamaki, J. I. J. Electrochem.Soc. 2011, 158, A1067. doi: 10.1149/1.3611434
(97) Arun, N.; Aravindan, V.; Ling, W. C.; Madhavi, S. J. Alloys Compd.2014, 603, 48. doi: 10.1016/j.jallcom.2014.03.059
(98) Mohamed, A. I.; Whitacre. J. F. Electrochim. Acta 2017, 235, 730.doi: 10.1016/j.electacta.2017.03.106
(99) Wu, W.; Mohamed, A.; Whitacre, J. F. J. Electrochem. Soc. 2013,160, A497. doi: 10.1149/2.054303jes
(100) Wu, W.; Yan, J.; Wise, A.; Rutt, A.; Whitacre, J. F. J. Electrochem.Soc. 2014, 161, A561. doi: 10.1149/2.059404jes
(101) Pang, G.; Nie, P.; Yuan, C. Z.; Shen, L. F.; Zhang, X. G.; Zhu, J. J.;Ding, B. Energy Technol. 2014, 2, 705.doi: 10.1002/ente.201402045
(102) Li, X. N.; Zhu, X. B.; Liang, J. W.; Hou, Z. G.; Wang, Y.; Lin, N.;Zhu, Y. C.; Qian, Y. T. J. Electrochem. Soc. 2014, 161, A1181.doi: 10.1149/2.0081409jes
(103) Zhao, B. D.; Lin, B.; Zhang, S.; Deng, C. Nanoscale 2015, 7,18552. doi: 10.1039/C5NR06505d
(104) Hung, T. F.; Lan, W. H.; Yeh, Y. W.; Chang, W. S.; Yang, C. C.; Lin,J. C. ACS Sustain. Chem. Eng. 2016, 4, 7074.doi: 10.1021/acssuschemeng.6b01962
(105) He, Y. W.; Yuan, H.; Wu, Y. X.; Chen, C.; Yang, S.; Ai, C. C.Electrochemistry 2016, 84, 705.doi: 10.5796/electrochemistry.84.705
(106) Ke, L. L.; Dong, J.; Lin, B.; Yu, T. T.; Wang, H. F.; Zhang, S.;Deng, C. Nanoscale 2017, 9, 4183. doi: 10.1039/C7NR00793K
(107) Minakshi, M.; Ralph, D. ECS Trans. 2013, 45, 95.doi: 10.1149/04529.0095ecst
(108) Pasta, M.; Wessells, C. D.; Liu, N.; Nelson, J.; McDowell, M. T.;Huggins, R. A.; Toney, M. F.; Cui, Y. Nat. Commun. 2014, 5, 3007.doi: 10.1038/ncomms4007
(109) Choi, W.; Harada, D.; Oyaizu, K.; Nishide, H. J. Am. Chem. Soc.2011, 133, 19839. doi: 10.1021/ja206961t
(110) Liang, Y. L.; Jing, Y.; Gheytani, S.; Lee, K. Y.; Liu, P.; Facchetti,A.; Yao, Y. Nat. Mater. 2017, 16, 841. doi: 10.1038/nmat4919
(111) Liu, Y.; Qiao, Y.; Zhang, W. X.; Wang, H.; Chen, H. K.; Zhu, H. P.;Li, Z.; Huang, Y. H. J. Mater. Chem. A 2015, 3, 7780.doi: 10.1039/C5TA00396B
(112) Li, Z.; Young, D.; Xiang, K.; Carter, W. C.; Chiang, Y. M. Adv.Energy Mater. 2013, 3, 290. doi: 10.1002/aenm.201200598
(113) Zhang, Q.; Liao, C. Y.; Zhai, T. Y.; Li, H. Q. Electrochim. Acta 2016, 196, 470. doi:10.1016/j.electacta.2016.03.007
(114) Whitacre, J. F.; Wiley, T.; Shanbhag, S.; Wenzhuo, Y.; Mohamed,A.; Chun, S. E.; Weber, E.; Blackwood, D.; Lynch-Bell, E.;Gulakowski, J.; et al. J. Power Sources 2012, 213, 255.doi: 10.1016/j.jpowsour.2012.04.018
(115) Liu, Y.; Zhang, B. H.; Xiao, S. Y.; Liu, L. L.; Wen, Z. B.; Wu, Y. P.Electrochim. Acta 2014, 116, 512.doi: 10.1016/j.electacta.2013.11.077
(116) Minakshi, M.; Meyrick, D. Electrochim. Acta 2013, 101, 66.doi: 10.1016/j.electacta.2013.02.075
(117) Hou, Z. G.; Li, X. N.; Liang, J. W.; Zhu, Y. C.; Qian, Y. T. J. Mater.Chem. A 2015, 3, 1400. doi: 10.1039/C4TA06018K
(118) Qu, Q. T.; Shi , Y.; Tian, S.; Chen, Y. H.; Wu, Y. P.; Holze, R.;J. Power Sources 2009, 194, 1222.doi: 10.1016/j.jpowsour.2009.06.068
(119) Zhang, B. H.; Liu, Y.; Wu, X. W.; Yang, Y. Q.; Chang, Z.; Wen, Z.B.; Wu, Y. P. Chem. Commun. 2014, 50, 1209.doi: 10.1039/c3cc48382g
(120) Wang, H., Zhang, T., Chen, C.; Ling, M.; Lin, Z.; Zhang, S. Q.;Pan, F.; Liang, C. D. Nano Res. 2017, doi:10.1007/s12274-017-1657-5
(121) Gao, H. C.; Goodenough, J. B. Angew. Chem. Int. Ed. 2016, 128,12960. doi:10.1002/ange.201606508
