SMRT测序技术及其在微生物研究中的应用
2018-06-29唐勇刘旭
唐勇 刘旭
(1. 乐山职业技术学院,乐山 614000;2. 乐山丰野农业科技有限责任公司,乐山 614000;3. 乐山市农业局,乐山 614000)
以Sanger测序法[1]为代表的第一代测序技术为分子生物学研究带来一场彻底的变革。Sanger测序技术已经为分子生物学研究服务近40年,其为科学研究所作出的贡献有目共睹。尽管第一代测序技术有着其不可替代的优势,但是其低通量的缺陷终究无法完全满足研究需要。21世纪,测序技术发展进入快车道,第二代测序技术[2]和第三代测序技术[3]相继问世。以 Roche/454[4]、Illumina/Solexa[5]等测序平台为代表的第二代测序技术解决测序通量和测序价格问题,引起生命科学研究方法大变革[6],但是,第二代测序技术也遗留下测序读长短的缺陷[7]。因此,为解决读长问题而发明的第三代测序技术应运而生[3]。
目前主流的第三代测序技术主要包括牛津纳米孔公司(Oxford Nanopore)的单分子纳米孔测序技术(The single-molecule nanopore DNA sequencing)、Helicos公司的真正单分子测序技术(True singlemolecule sequencing,tSMS) 和 Pacific Biosciences(PacBio)公司的单分子实时测序技术(Single-molecule real-time,SMRT)[8]。其中,牛津纳米孔技术有限公司开发的单分子纳米孔测序技术以超长读长和轻便见长[9],然而,由于其测序错误率高达35%[10-11]无法在研究中推广;Helicos公司的tSMS测序技术费用偏高[12],项目基本处于停滞状态。目前,最成熟的第三代测序平台莫过于基于SMRT测序技术的PacBio系列平台。
测序技术的发展对微生物研究的推动作用明显,尤其是不可培养的微生物和复杂环境微生物的研究[13-14]。目前,微生物研究依然以第二代测序技术为主。但是,随着基于SMRT测序技术的PacBio系列测序平台的进一步成熟,其必将成为微生物研究者手中的另一柄利剑。因此,系统了解SMRT测序技术的特点及其在微生物研究中的应用进展,对微生物研究者具有指导意义。本文将介绍SMRT测序技术的原理和特点,详细列举SMRT测序技术在微生物16S rRNA基因全长测序、宏基因组测序和微生物全基因组测序中的应用,以及下游分析中存在的问题,旨为使用SMRT测序技术研究微生物的研究人员提供一定参考。
1 SMRT测序技术原理
和其他两个单分子测序技术原理一样,SMRT测序技术也采用边合成边测序的策略。SMRT测序技术的核心是零模波导孔(Zero mode waveguide,ZMW),ZMW是直径20-50纳米的纳米孔,底部固定有DNA聚合酶[15]。数百纳米的激光照着DNA聚合酶所在的ZMW底部而发生衍射照亮狭小的范围,从而刚好检测到进入ZMW底部的碱基所携带荧光基团,而避免背景干扰(图1-A[16])。每个ZMW可以结合一个DNA模板,其测序过程(图1-B[16])是:(1)DNA聚合酶捕获DNA单链模板并结合在活性位点上;(2)被不同染料标记的脱氧核苷酸进入ZMW底部检测区与聚合酶结合;(3)基于脱氧核苷酸在ZMW底部停留时间判断是否匹配;(4)被标记的磷酸基团被切割并释放[12]。
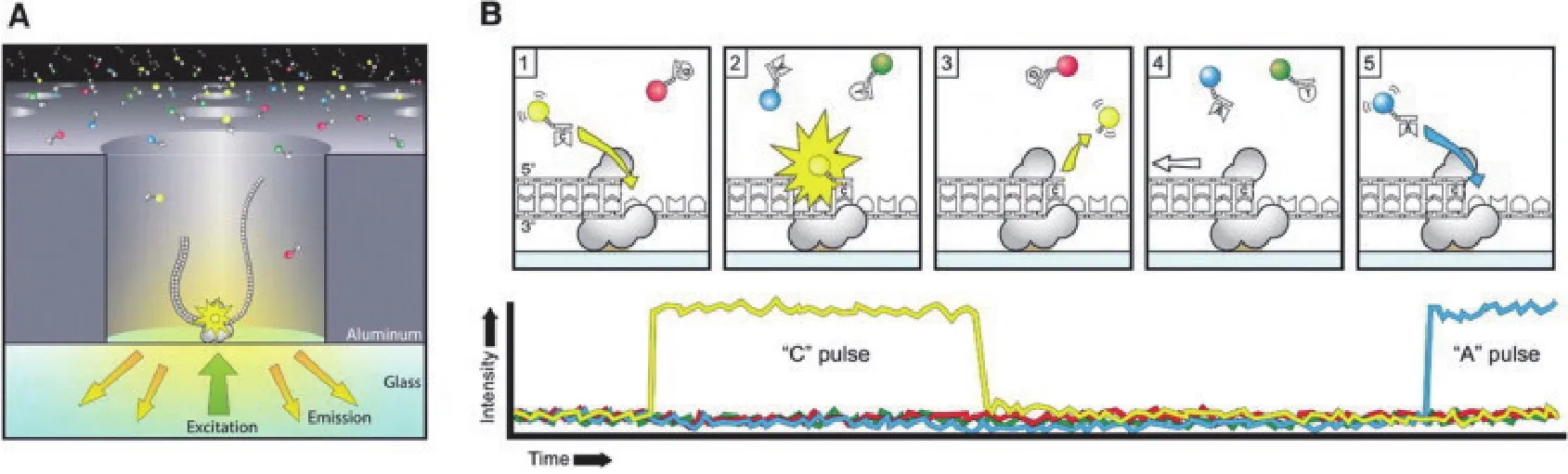
图1 SMRT测序技术原理[16]
2 SMRT测序技术特点
作为第三代测序的基本特点,测序长度是SMRT测序技术的优势之一。Ferrarini等[17]使用PacBio RS平台,P4/C2试剂对叶绿体基因组进行测序,结果获得平均reads长度为3 936.66 bp,一致序列的平均碱基错误率为1.3%。Shearman等[18]使用最新的PacBio SR Ⅱ平台和C4试剂测序,成功获得长度大于26 kb的reads。理论上,在最新的PacBio SR Ⅱ平台下,使用P6/C4试剂测序能够获得的最长reads可以达到60 kb[16]。由于DNA聚合酶在激光的照射下会逐渐失活,因此其测序长度不可能永远增加[19]。
测序错误偏高是所有测序技术都面临的问题。基于纳米孔测序技术的MinION测序仪和基于SMRT测序技术的PacBio平台测序reads错误率分别达到 40% 和 15%[11,20]。然而,PacBio测序平台所产生的测序错误为随机分布的单碱基错误、插入或缺失[20-21],凭借这一特点,PacBio引入环化测序的策略成功将测序准确度提高,即将双链模板两端加载发夹结构接头,形成环状的测序模板(SMRTbell),然后对模板循环测序[22]。该测序方案可以保证相同碱基被多次测序,结合错误随机模型,采用多重比对可以修正错误碱基,从而获得高准确度reads[23]。该方案在全长16S rRNA基因测序、转录组测序等对reads长度要求相对较低,但是对测序准确度要求较高的研究中非常有效[24-25]。
测序速度快是SMRT测序技术的另一特点。相比动辄运行数天的第二代测序技术,SMRT测序技术每个run运行时间最短近0.5 h[16]。虽然,每个run输出的数据量远远不及Hiseq2500等第二代测序技术,但是在对时间要求较高的情况下,SMRT测序技术无疑极具优势(表1),如在临床检测或者疫情爆发等情况下。
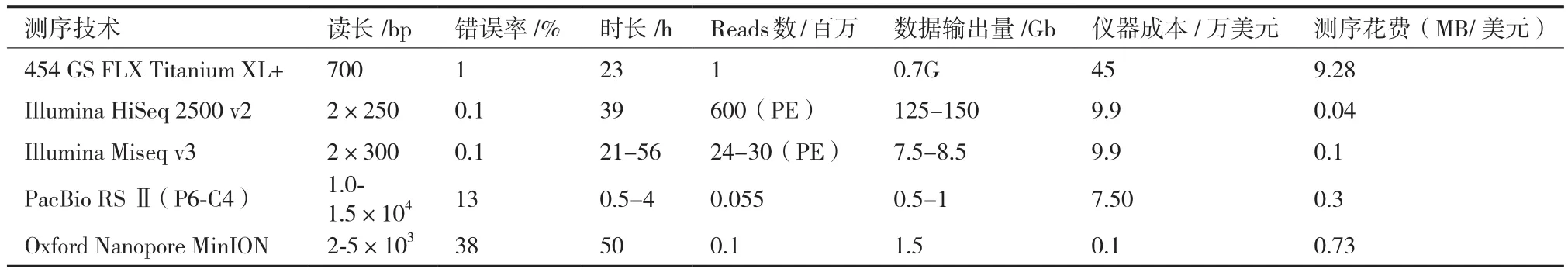
表1 主要高通量测序仪器参数[16,26-28]
3 SMRT测序技术在微生物研究中的应用
3.1 微生物16S rRNA基因测序
自2006年,Sogin等[29]首次成功将高通量测序技术(罗氏454)用于深海环境微生物多样性调查,16S rRNA基因高通量测序片段选择一直存在争议[30],全长DNA测序无疑可以彻底终止这一争论。SMRT测序技术在复杂环境微生物的研究中所具备的优势已经被多次证实[24,31]。随着SMRT测序技术的技术成熟和测序成本降低,第三代测序技术在16S rRNA基因测序中的应用越来越多。
肠道微生物与宿主的生长、免疫和健康息息相关,对肠道微生物调查有利于对相关疾病的标记与治疗。2013年,Hu等[32]采集23个采用不同分娩方式出生的新生儿的粪便(10个孩子母亲患有糖尿病和13个孩子母亲未患糖尿病),采用PacBio RS平台测序粪便中16S rRNA基因的V3-V4区,分析PASS数大于3的CCS reads,结果得到与其他实验相反的结果:不同分娩方式对新生儿的粪便微生物没有影响,而母亲患病状态对新生儿肠道微生物组成有显著的影响。泡菜中含有大量乳酸菌和其他杂菌,四川家庭自制泡菜微生物的组成并不清楚。2017年,Cao等[33]在重庆7个地区采集到38份10年以上的泡菜盐水,通过滴定法分为高酸度、中等酸度和低酸度3组。采用SMRT测序技术(PacBio SR Ⅱ/P6-C5)对38个样本的16S rRNA基因全长进行测序。通过分析聚类和注释分析得来自371个属的593个种(包括35个门),其中,Lactobacillus acetotolerans的丰度与酸度呈正相关。此外,Serratia marcescens和Stenotrophomonas maltophilia等机会致病菌也在样本中检测到。酸度越低,物种多样性越高,乳酸菌属内的菌种越多(丰度大于1%),机会致病菌越多。该研究为自制泡菜的进一步研究提供了参考,且表明需要对自制四川泡菜内的机会致病菌深入研究。
3.2 宏基因组测序
宏基因组是指环境中的所有微生物基因组的总和[34-35]。2000年,Rondon等[35]首次通过构建宏基因组文库研究土壤微生物多样性,并开启了环境微生物研究的新篇章。随着高通量测序价格大幅下跌,获得大批原始宏基因组测序数据已经不再是难题,而真正的研究瓶颈在于数据分析环节。其中,微生物参考基因组缺乏是宏基因组数据分析主要障碍。目前,已有参考基因组的微生物数量与自然界存在的微生物数量相去甚远(表2)。因此,从复杂的宏基因组数据中完整而准确地构建微生物基因组草图成为分析流程的首要任务[36]。第二代测序技术由于测序片段短的问题导致组装困难,第三代测序技术有望彻底解决这一问题。
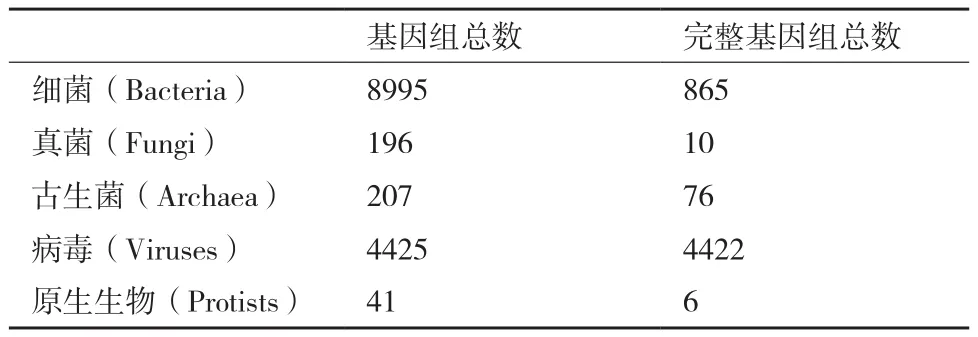
表2 微生物参考基因组统计
2016年,Frank等[37]采用Hiseq2000和PacBio RS Ⅱ两种平台结合的测序方式对沼气反应器内的微生物宏基因组进行研究。其中,构建插入片段为1.5 kb的SMRTbell文库使用P4/C2试剂测序。分别单独组装两份数据,再采用混合组装的方式组装。结果表明,混合组装的方式得到的组装序列长度高于单独组装。该试验结果表明SMRT测序技术对微生物宏基因组研究有提高作用。Frank等采用混合的方式是考虑SMRT测序技术测序成本(深度)的问题,而采用较为折中的方案。事实上,随着PacBio系列测序平台的普及和价格快速下降,SMRT可以完全取代第二代测序。2017年,Driscoll等[38]从美国克拉马斯湖中采集水样并共培养。然后采用PacBio测序平台进行宏基因组测序并得到348 623条平均长度达到7 737 bp的PacBio reads,经过质量过滤和组装,他们发现成功组装出3个微生物基因组草图。Driscoll等的实验证明SMRT测序技术在低复杂度环境微生物宏基因组组装中是有效的。
3.3 微生物全基因组测序
对无参考基因组的物种,采用测序并从头组装获得全基因组图谱的方式称为全基因组测序。SMRT测序技术测序长度能够帮助研究人员在组装全基因组时成功跨过重复片段、低复杂区域,从而组装出完整性更好的全基因组[39]。2013年,Chin等[40]设计并开发针对SMRT测序数据组装微生物全基因组的算法(HGAP),他们使用该方法成功组装了包括大肠杆菌(E. coli)在内的16个基因组,其中,3个基因组已经有完整的基因组,新组装的基因组与参考基因组一致性达到99.9999%。他们的实验证明结合SMRT测序技术和Illumina测序技术进行全基因组测序准确有效。Paulinella chromatophora是研究植物质体的重要模式生物,2017年,Lhee等[41]研究发现一个具有光合作用的新种(P. micropora sp.nov.),通过构建SMRTbell库,并使用PacBio RS II测序平台测序获得16 Gb数据,使用HGAP算法组装得到长度为976 991 bp的全基因组。通过全基因组水平的比较证实其为新的种。
除了完全采用PacBio reads进行全基因组组装,通过与第二代测序技术组合的方式也是常用的微生物全基因组组装方案,该方法能够有效提高组装准确性并降低测序成本。葡萄孢菌(Botrytis cinerea)是广泛存在的植物病原真菌,研究人员先后使用第一代和第二代测序技术对全基因组测序,但是,其中仍然存在较多缺失和错误,2016年,Van Kan等[42]采用SMRT测序技术和第二代测序技术结合的方式对葡萄孢菌全基因组测序,de novo组装得到由18条染色体组装的新基因组,测序深度和完整性得到大幅提高。同时,他们采用RNAseq数据对基因组进行验证和基因注释。
4 结语
微生物物种数量庞大,而环境微生物复杂性决定了其对研究技术的高要求。尽管第二代测序技术为微生物研究带来了革命性的改变,但是,以SMRT测序技术为代表的第三代测序技术取代第二代测序技术成为微生物研究的主要手段是必然趋势。SMRT测序技术已然领跑第三代测序技术。但是,SMRT测序技术仍然存在较大的问题,如测序费用高、测序错误率偏高等。
目前,SMRT测序技术在微生物研究领域应用最成熟且最多的还是微生物全基因组测序。而SMRT测序技术在复杂环境微生物宏基因组研究中还存在诸多问题需要解决,已有的研究也只是浅尝辄止。目前,在我们文献查阅的范围之内,还没有发现真正将SMRT测序技术应用于复杂环境微生物研究,因此,这方面还需要进一步探索。SMRTbell库构建方法的提出为SMRT技术在微生物16S rRNA基因全长测序提供了可能,最近两年逐渐在研究中被采用。但是,目前16S rRNA基因注释数据库还存在注释物种少,参考序列长度短的问题,这无疑将降低16S rRNA基因全长测序数据分析的准确性。
[1]Sanger F, Nicklen S, Coulson AR. DNA sequencing with chainterminating inhibitors[J]. Proc Natl Acad Sci USA, 1977, 74(12):5463-5467.
[2]Metzker ML. Sequencing technologies—the next generation[J].Nature Reviews Genetics, 2010, 11(1):31-46.
[3]Schadt EE, Turner S, Kasarskis A. A window into third-generation sequencing[J]. Human Molecular Genetics, 2011, 19(4):R227-R240.
[4]Margulies M, Egholm M, Altman WE, et al. Genome sequencing in microfabricated high-density picolitre reactors[J]. Nature, 2006,437(7057):376-380.
[5]Bentley DR. Whole-genome re-sequencing[J]. Current Opinion in Genetics & Development, 2006, 16(6):545-552.
[6]Reis-Filho JS. Next-generation sequencing[J]. Breast Cancer Research, 2009, 11(3):S12.
[7]Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing:computational challenges and solutions[J]. Nature Reviews Genetics, 2012, 13(1):36-46.
[8]柳延虎, 王璐, 于黎. 单分子实时测序技术的原理与应用[J].遗传, 2015, 37(3):259-268.
[9]Clarke J, Wu HC, Jayasinghe L, et al. Continuous base identification for single-molecule nanopore DNA sequencing[J]. Nature Nanotechnology, 2009, 4(4):265-270.
[10]Goodwin S, Gurtowski J, Ethe-Sayers S, et al. Oxford nanopore sequencing, hybrid error correction, and De novo assembly of a eukaryotic genome[J]. Biorxiv, 2015, 25(11):1750-1756.
[11]Laver T, Harrison J, O’Neill PA, et al. Assessing the performance of the oxford nanopore technologies minion[J]. Biomolecular Detection & Quantification, 2015, 3:1-8.
[12]Treffer R, Deckert V. Recent advances in single-molecule sequencing[J]. Current Opinion in Biotechnology, 2010, 21(1):4-11.
[13]Xia W, Jia Z. Comparative analysis of soil microbial communities by pyrosequencing and dgge[J]. Acta microbiologica Sinica,2014, 54(12):1489-1499.
[14]Shokralla S, Spall JL, Gibson JF, et al. Next-generation sequencing technologies for environmental DNA research[J]. Molecular Ecology, 2012, 21(8):1794-1805.
[15]Levene MJ, Korlach J, Turner SW, et al. Zero-mode waveguides for single-molecule analysis at high concentrations[J]. Annual Review of Biophysics, 2012, 41(41):269-293.
[16]Rhoads A, Au KF. Pacbio sequencing and its applications[J].Genomics, Proteomics & Bioinformatics, 2015, 13(5):278-289.
[17]Ferrarini M, Moretto M, Ward JA, et al. An evaluation of the pacbio rs platform for sequencing and De novo assembly of a chloroplast genome[J]. BMC Genomics, 2013, 14(1):670.
[18]Shearman JR, Sonthirod C, Naktang C, et al. The two chromosomes of the mitochondrial genome of a sugarcane cultivar:assembly and recombination analysis using long pacbio reads[J]. Scientific Reports, 2016, 6:31533.
[19]李明爽, 赵敏. 第三代测序基本原理[J]. 现代生物医学进展,2012, 12(10):1980-1982.
[20]Koren S, Schatz MC, Walenz BP, et al. Hybrid error correction de novo assembly of single-molecule sequencing reads[J]. Nature Biotechnology, 2012, 30(7):693-700.
[21]Ross MG, Russ C, Costello M, et al. Characterizing and measuring bias in sequence data[J]. Genome Biology, 2013, 14(5):R51.
[22]Kong N, Thao K, Ng W, et al. Automation of PacBio SMRTbell 10 Kb template preparation on an agilent NGS workstation[J]. Crop Science, 2014, 15(6):886.
[23]Eid J, Fehr A, Gray J, et al. Real-time DNA sequencing from single polymerase molecules[J]. Methods in Enzymology, 2009, 323(5910):133.
[24]Schloss PD, Jenior ML, Koumpouras CC, et al. Sequencing 16S RNA gene fragments using the PacBio SMRT DNA sequencing system[J]. Peerj, 2015, 4:e1869.
[25]Gao S, Ren Y, Sun Y, et al. PacBio full-length transcriptome profiling of insect mitochondrial gene expression[J]. RNA Biology, 2016, 13(9):820-825.
[26]Goodwin S, McPherson JD, McCombie WR. Coming of age:ten years of next-generation sequencing technologies[J]. Nature Reviews Genetics, 2016, 17(6):333-351.
[27]Giordano F, Aigrain L, Quail MA, et al. De novo yeast genome assemblies from MinION, PacBio and MiSeq platforms[J].Scientific reports, 2017, 7(1):3935.
[28]Mikheyev AS, Tin MM. A first look at the Oxford Nanopore MinION sequencer[J]. Molecular Ecology Resources, 2014, 14(6):1097-1102.
[29]Sogin ML, Morrison HG, Huber JA, et al. Microbial diversity in the deep sea and the underexplored “rare biosphere”[J].Proceedings of the National Academy of Sciences, 2006, 103(32):12115-12120.
[30]Chakravorty S, Helb D, Burday M, et al. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria[J]. Journal of Microbiological Methods, 2007, 69(2):330-339.
[31]Mosher JJ, Bowman B, Bernberg EL, et al. Improved performance of the PacBio SMRT technology for 16S rDNA sequencing[J].Journal of Microbiological Methods, 2014, 104:59-60.
[32]Hu J, Nomura Y, Bashir A, et al. Diversified microbiota of meconium is affected by maternal diabetes status[J]. PLoS One,2013, 8(11):e78257.
[33]Cao J, Yang J, Hou Q, et al. Assessment of bacterial profiles in aged, home-made Sichuan paocai brine with varying titratable acidity by PacBio SMRT sequencing technology[J]. Food Control, 2017, 78:14-23.
[34]Handelsman J, Rondon MR, Brady SF, et al. Molecular biological access to the chemistry of unknown soil microbes:a new frontier for natural products[J]. Chemistry &Biology, 1998, 5(10):R245-R249.
[35]Rondon MR, August PR, Bettermann AD, et al. Cloning the soil metagenome:a strategy for accessing the genetic and functional diversity of uncultured microorganisms[J]. Applied and Environmental Microbiology, 2000, 66(6):2541-2547.
[36]Howe A, Chain PS. Challenges and opportunities in understanding microbial communities with metagenome assembly(Accompanied by Ipython Notebook Tutorial)[J]. Frontiers Microbiol, 2015, 6:678.
[37]Frank JA, Pan Y, Toomingklunderud A, et al. Improved metagenome assemblies and taxonomic binning using long-read circular consensus sequence data[J]. Scientific Reports, 2016, 6:25373.
[38]Driscoll CB, Otten TG, Brown NM, et al. Towards long-read metagenomics:complete assembly of three novel genomes from bacteria dependent on a diazotrophic cyanobacterium in a freshwater lake Co-culture[J]. Stand Genomic Sci, 2017, 12:9.
[39]Powers JG, Weigman VJ, Shu J, et al. Efficient and accurate whole genome assembly and methylome profiling of E. coli[J]. BMC Genomics, 2013, 14(1):675.
[40]Chin CS, Alexander DH, Marks P, et al. Nonhybrid, finished microbial genome assemblies from long-read smrt sequencing data[J]. Nature Methods, 2013, 10(6):563-569.
[41]Lhee D, Yang EC, Im Kim J, et al. Diversity of the photosynthetic Paulinella species, with the description of Paulinella micropora sp.nov. and the chromatophore genome sequence for strain Kr01[J].Protist, 2017, 168(2):155-170.
[42]Van Kan JA, Stassen JH, Mosbach A, et al. A gapless genome sequence of the fungus botrytis cinerea[J]. Mol Plant Pathol,2017, 18(1):75-89.
