高效液相色谱-荧光检测法测定华法林钠片的含量
2018-06-28
华法林钠是一种双香豆素类抗凝药物[1],其能抑制肝脏内凝血酶原和凝血因子合成。该药物口服于胃肠道吸收迅速而完全,临床上常制成片剂用于辅助治疗肺栓塞、术后栓塞、心肌梗死等血栓栓塞性疾病[2]。华法林钠用量少,规格通常为每片2.5 mg,过量使用易致各种出血,针对肝肾功能不全、活动性溃疡、严重高血压、脑出血患者需慎用[3]。因此,需要灵敏、准确的测定方法作为华法林钠片质量控制的有效方法[4]。近年来,国内外学者对测定华法林钠含量的方法进行了较多研究[5],但对采用高效液相色谱(HPLC)-荧光检测法测定华法林钠片的含量却鲜有报道。HPLC-荧光检测法灵敏,简便,快捷。本研究就 HPLC-荧光检测法测定华法林钠片的含量,现报道如下。
1 仪器与试剂
1.1 仪器高效液相色谱仪(日本岛津公司);HW-2000色谱工作站;AY120D电子天平(日本岛津公司);UV1101紫外-可见分光光度计(上海天美科学仪器有限公司);DL-360超声波清洗仪;HP-01溶剂过滤器;ELGA超纯水仪。
1.2 试剂与试药华法林钠片(上海信宜九福药业有限公司,规格:2.5 mg/片,批号:080701、070603、080901);华法林钠化学原料药(由上海信宜九福药业有限公司提供,纯度99.2%);所用试剂甲醇为色谱纯,其余均为分析纯,水为超纯水。
2 方法与结果
2.1 溶液配制
2.1.1 对照溶液配制精密称取华法林钠化学原料药50.7 mg,置于50 ml容量瓶中,加适量甲醇至刻度,摇匀,配制成1.006 mg/ml的华法林钠对照贮备溶液[6]。精密量取5 ml,置于25 ml容量瓶中,加甲醇至刻度,摇匀,精密量取2 ml,置于25 ml容量瓶中,加流动相至刻度,摇匀,配制成16.1 μg/ml的华法林钠对照溶液。
2.1.2 供试品溶液配制取样品华法林钠20片,精密称定,研细,混匀。精密称取适量(约相当于华法林钠5.0 mg),置于25 ml容量瓶中,加甲醇约15 ml,超声 10 min,搅拌使其充分溶解,加甲醇至刻度,摇匀,滤过,用移液瓶量取续滤液2 ml,置于25 ml容量瓶中,加流动相至刻度,摇匀,用滤膜过滤,配制成供试品溶液。
2.1.3 阴性空白对照溶液按中国药典处方自制空白片,精密称取适量,按2.1.2项下供试品溶液配制方法配制成阴性空白对照溶液[7]。
2.2 色谱条件及系统适用性试验色谱柱:Spherisorb C18(4.6 mm×200 mm,10 μm);流动相:甲醇:0.1%磷酸(75:25);流速:1.0 ml/min;柱温:室温;检测波长:激发波长280 nm,发射波长380 nm。不同波长条件的测定结果以及 HPLC-荧光检测法和紫外-分光光度法(中国药典 2015版)测定[8-9]样品结果见表1-2。分别取华法林钠对照溶液、供试品溶液和阴性空白对照溶液20 μl,按上述色谱条件,注入液相色谱仪,记录色谱图(见图1-3)。在上述色谱条件下,理论板数以华法林钠计算不低于 2800,保留时间约为4.8 min。华法林钠阴性空白对照溶液图谱显示在主峰保留时间前后基线平稳,可见辅料对主药测定无干扰[10]。

表1 不同波长条件的测定
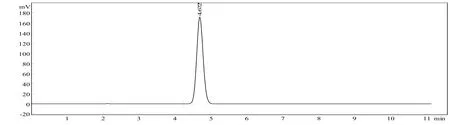
图1 对照溶液HPLC图

图2 供试品溶液HPLC图谱
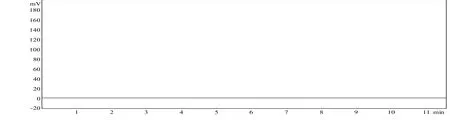
图3 阴性空白对照溶液HPLC图谱

表2 两种方法测定样品结果比较(n=3)
2.3 方法学考察
2.3.1 标准曲线绘制精密量取1.006 mg/ml的华法林钠对照贮备溶液5 ml,置于25 ml容量瓶中,加甲醇至刻度,充分摇匀,得201 μg/ml溶液,分别用量瓶量取上述溶液 0.5、1.0、2.0、3.0、4.0、5.0 ml,置于25 ml容量瓶中,加流动相至刻度,摇匀,即得浓度分别为4.03、8.06、16.1、24.2、32.2、40.3 μg/ml的华法林钠对照溶液。按色谱条件进行测定,结果见表3。以峰面积(Y)为纵坐标,华法林钠对照溶液浓度(X)为横坐标绘制标准曲线,其回归方程为:Y=130 952X+65 016,相关系数r=0.9999(n=6),结果见图4。结果表明,华法林钠在4.03~40.3 μg/ml浓度范围内,峰面积与其浓度呈良好线性关系[11-12]。

表3 标准曲线测定的结果
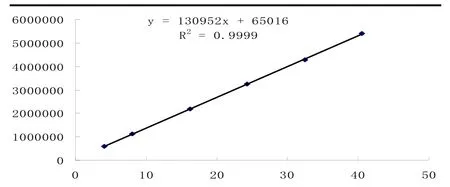
图4 华法林钠峰面积与华法林钠浓度线性关系图
2.3.2 精密度试验按色谱条件下,取浓度为16.1 μg/ml的华法林钠对照溶液,进样20 μl,连续重复进样6次,记录峰面积,华法林钠对照溶液峰面积的相对标准偏差(RSD)为0.60%(n=6),表明此仪器精密度良好。结果见表4。
2.3.3 稳定性试验取新配制的供试品溶液,分别于 0、2、4、6、8 、12、24 h 进样 20 μl,记录色谱图,测定华法林钠峰面积,峰面积RSD为1.9%,可见华法林钠供试品溶液在2 h内稳定。结果见表5。
2.3.4 重复性试验精密称取同一批号样品 6份,按“3.1.2”项下制备方法制备供试品溶液,进样20 μl,测得相对标示含量的RSD为0.71%(n=6)。表明此方法的重复性良好,结果见表6。

表4 精密度试验结果
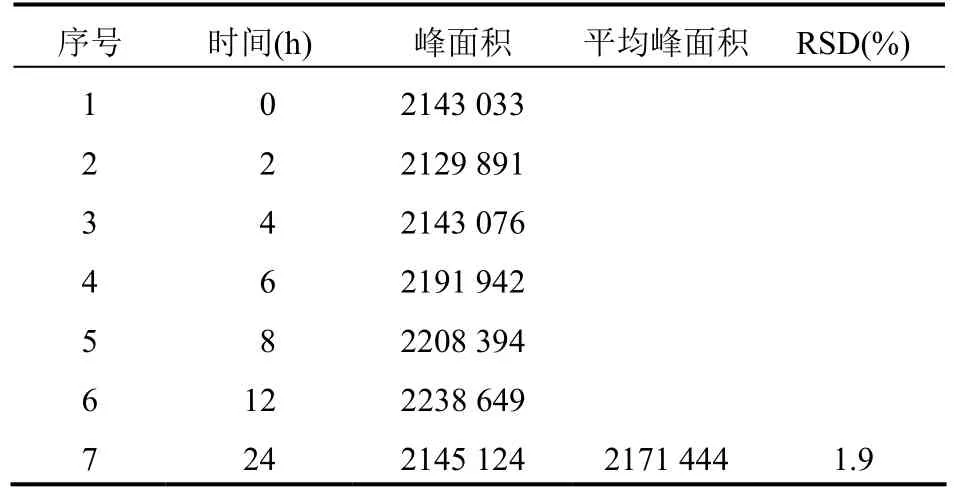
表5 稳定性试验结果

表6 重复性试验结果
2.3.5 回收率试验取华法林钠对照品适量,精密称定。分别按处方加入辅料,按“3.1.2”项下方法制备供试品溶液。分别进样20 μl,测定,记录峰面积。结果平均回收率为101.1%,RSD=0.60%(n=6),表明此方法准确可靠,见表7。
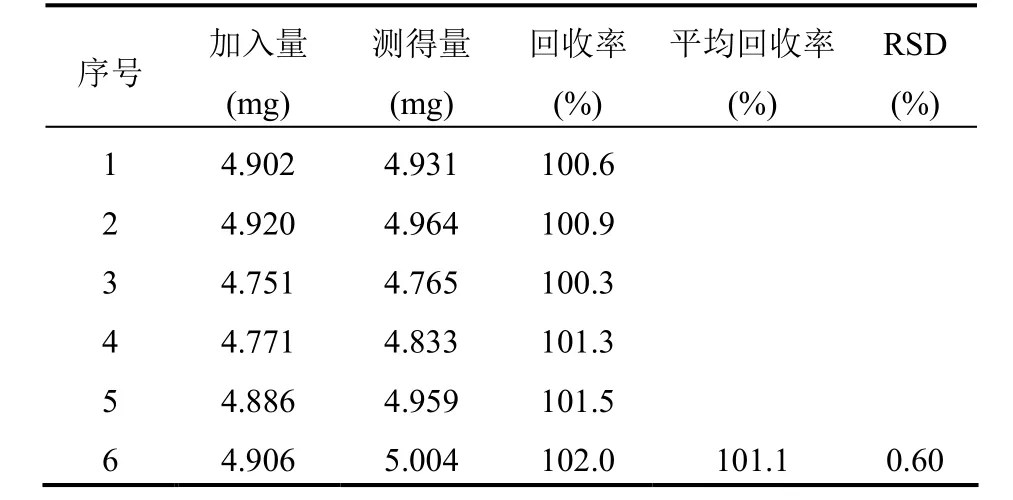
表7 回收率试验结果
2.3.6 样品测定分别取 3个批号的华法林钠片,精密称取适量,按照“3.1.2”项下方法制备供试品溶液,按色谱条件进行测定,测得相对标示百分含量分别为101.3%、101.5%和101.7%,结果见表8。

表8 样品测定结果
3 讨论
3.1 HPLC流动相选择参考相关研究报道的华法林及其衍生物采用的流动相,并根据实际情况,进行流动相选择试验,采用不同比例甲醇:1%冰醋酸、甲醇:0.2%冰醋酸、甲醇:水、0.1%磷酸:0.15%三已胺:已腈、甲醇:0.1%磷酸进行试验,结果表明流动相为甲醇:0.1%磷酸(75:25)时,华法林钠色谱峰形好,分离度符合要求,灵敏度高,保留时间适中[13]。因此选用甲醇:0.1%磷酸(75:25)为流动相,流速为1.0 ml/min。
3.2 检测波长选择研究报道[14]华法林及其衍生物发射波长在380 nm处有最大吸收,而最大激发波长为270~310 nm。取对照溶液20 ul,分别在激发波长为270、280、290、300、310 nm及发射波长为380 nm时进样,结果以280 nm作为激发波长、380 nm作为发射波长,峰面积最大,灵敏度较高。因此,选用280 nm作为激发波长、380 nm作为发射波长。
3.3 实验过程需要注意的问题在实验过程中使用的玻璃仪器需采用洗涤剂冲洗干净,防止污染荧光物质,不能用洗衣粉清洗仪器,因为其含有荧光增白剂,会对测定样品造成严重干扰[15];而测定样品中的辅料不会对荧光测定有影响,华法林钠的荧光对温度很敏感,温度越低,荧光越灵敏,测出的峰面积越大,在测试过程应保持温度恒定,恒定温度可确保实验测定结果稳定[16]。
3.4 紫外-分光光度计算法与 HPLC-荧光检测法比较中华人民共和国药典(2015年版)采用紫外-分光光度法测定华法林钠片含量,样品需采用三氯甲烷和0.01 mol/L的NaOH分别提取3次,耗时较长[17]。而采用 HPLC-荧光检测法测定华法林钠片含量,样品直接加甲醇超声10 min即可,方法简便、快捷、灵敏,且与外-分光光度法测定的结果相一致。
4 结论
药物分析的方法应该不断探索,寻找更简单、更准确的方法,需要敢于创新,能更有效用于药物质量控制。本研究结果表明,采用 HPLC-荧光检测法测定华法林钠片含量方法简便、重复性好,其测定的华法林钠片含量为4.03~40.3μg/ml,结果准确,重现性好,可作为华法林钠片质量控制的有效方法[5,17]。
[1]李瑞,殷明.药理学[M].6 版.北京:人民出版社,2007.
[2]王宪德,周文秀,刘苏,等.华法林血浆浓度的测定在抗凝监测中的意义[J].河北医科大学学报,2003,24(4):193-196.
[3]陈德,王俊,王鸣和.华法林研究进展[J].世界临床药物,2005,26(6):375-378.
[4]李发美.医药高效液相色谱技术[M].北京:人民卫生出版社,1999.
[5]盛龙生,何丽一,徐连连,等.药物分析[M].北京:化学工业出版社,2002.
[6]Sastry CS,Rao TT,Sailaja A,et al.Micro-Determination of Warfarin Sodium,Nicoumalone and Acebutolol Hydroch Loride in Pharmaceutical Preparations[J].Talanta,1991,38(10):1107-1109.
[7]裴若会,闫宏涛,郭艳丽.抗凝药物华法林的荧光光谱测定方法研究[J].药物分析杂志,2008,28(1):140-142.
[8]中华人民共和国国家药典委员会.中华人民共和国药典(2015年版二部)[S].北京:化学工业出版社,2015.
[9]叶文鹏,刘俊亭,刁尧.二阶导数紫外光谱法测定华法林[J].理化检验(化学分册),2003,39(5):293-294,296.
[10]程司堃,张五龙,梅其炳.极谱法测定华法林钠的含量[J].解放军药学学报,2002,18(3):178-180.
[11]黄碧莹,姚秋燕,李敏薇,等.HPLC 法测定华法林片的含量[J].中国实用医药,2008,3(25):30-31.
[12]Ufer M,Kammerer B,Kirchheiner J,et al.Determination of Phenprocoumon,Warfarin and Their Monohydroxylated Metabolites in Human plasma and Urine by Liquid Chromatography-Mass spectrometry after solid-phase Extraction[J].J Chromatogr B Analyt Technol Biomed Life Sci,2004,809(2):217-226.
[13]李发美.医药高效液相色谱技术[M].北京:人民卫生出版社,1999:482-483.
[14]金米聪,陈晓红,李小平.高效液相色谱一荧光检测法同时测定全血中5种香豆素类杀鼠剂[J].色谱,2007,25(2):214-216.
[15]李发美.分析化学[M].5版.北京:人民卫生出版社,2003.
[16]Andersen C,Balmér K,Lagerstróm PO.Enantioselextive assay of warfrin in biood piasmaby Liquid chromatongraphy on Chiralcel OC[J].J Chromatogr,1993,615(1):159-463.
[17]郝润喜,阎彩萍,贺娟.高效液相色谱法测定华法令血药浓度[J].山西医科大学学报,1997,28(3):22-23.
