基于高通量测序的女贞果实转录组研究△
2018-06-25徐德宏罗月芳江灵敏孟蕾谭朝阳
徐德宏,罗月芳,江灵敏,孟蕾,谭朝阳
(湖南中医药大学 药学院 生物工程实验室,湖南 长沙 410208)
女贞LigustrumlucidumAit.为木犀科女贞属植物,其干燥成熟果实(即女贞子)是一种常用中药,具有滋补肝肾、明目乌发的功能,用于肝肾阴虚、眩晕耳鸣、腰膝酸软、须发早白、目暗不明、内热消渴、骨蒸潮热[1]。经化学成分分析和活性检测发现,女贞果实中含有多种具有药理活性的次级代谢物[2-3],如萜类、黄酮类等,因此近年来女贞果实在医药及保健方面备受人们的关注。目前,女贞果实的研究主要集中在次级代谢物的成分分析、提取和活性检测,而关于其功能基因发现和注释、物质合成相关途径分析、分子标记开发等转录组学方面的研究却未有报道,故现阶段有必要采用适当的方法对以上内容展开深入系统的研究。
近年来,各种组学研究相继兴起,其中第二代高通量测序技术特别是RNA测序技术已广泛应用于生物体转录组基因表达分析,采用该技术能全面快速地获取研究对象在某一状态下基因组转录信息,从中挖掘重要的功能基因,揭示不同生物学性状的分子机制[4-5]。Illumina/Solexa是目前转录组测序中最常使用的一种RNA高通量测序技术,与传统的EST sanger测序法相比,具有高通量、低成本和高灵敏度等无法比拟的优势,同时可对低丰度的基因表达进行检测[6]。此外,随着生物信息学的不断发展,特别是序列组装软件的开发,主要蛋白质数据库的更新和转录组分析方法的建立[7-9],为非模式生物进行转录组高通量测序、从头拼接组装和unigene的功能注释提供了极大的方便,因而Illumina/Solexa第二代高通量测序技术已广泛应用到了非模式生物的转录组研究中。
本研究将利用Illumina/Solexa Hiseq2000测序平台,对女贞果实进行转录组测序,将测序得到的大量数据进行拼接与组装,并应用生物信息学方法对所得序列进行功能注释、功能分类、代谢途径分析和简单重复序列(simple sequence repeat,SSR)位点查找等研究,从而在转录组水平系统研究女贞果实,并为进一步开展女贞果实的分子标记开发和重要次级代谢产物的生物合成奠定基础。
1 材料与方法
1.1 女贞果实转录组文库构建、测序和组装
女贞果实于2015年8月采摘于湖南中医药大学湘杏学院校区内的女贞树上,用Trizol试剂分别提取3棵女贞树成熟果实的总RNA,每个样本取等量混合组成RNA池,然后用磁珠富集含poly(A)的mRNA并将其打断成短序列,以这些短序列为模板用六碱基随机引物依次合成第1条cDNA链和第2条cDNA链。cDNA链经试剂盒纯化并加EB缓冲液洗脱之后做末端修复、加poly(A)并连接测序接头,然后用琼脂糖凝胶电泳选择200~500 bp片段进行PCR扩增,最后对建好的测序文库用Illumina/Solexa Hiseq2000测序平台进行测序。鉴于测序平台数据错误率对结果的影响,对原始reads数据进行质量预处理得到clean reads,即先使用cutadapt软件去除3′端测序接头[10];再利用滑动窗口法去除低质量片段:质量阈值20(错误率=1%),窗口大小10 bp,长度阈值35 bp;最后切除reads中含N部分序列:长度阈值35 bp。参照Grabherr 等提出方法[11],将所得clean reads通过Trinity软件(版本号trinityrnaseq_r2013-02-25)进行从头拼接组装,即通过clean reads之间的重叠信息组装得到重叠群(Contigs),然后局部组装得到转录本(Transcripts),最后用 Easycluster软件对Transcripts进行同源聚类以得到单基因簇(Unigene)[12-13]。
1.2 Unigene功能注释、GO分类和代谢路径分析
通过Blastx在e值<0.000 01的条件下,将Unigene序列比对到蛋白数据库Nr(Non-redundant protein database,非冗余蛋白数据库)、Swiss-Prot(SwissProt protein database,蛋白质序列数据库),并通过Blastn在e值<0.000 01的条件下,将Unigene比对到核酸数据库Nt(Nucleotide collection,核酸数据库),得到与给定Unigene具有最高序列相似性且功能已知的蛋白,从而实现Unigene的功能注释。根据Nr数据库的功能注释结果,使用Blast2GO软件得到unigene的GO条目[14],然后用WEGO软件对所有的unigene进行GO功能分类统计[15]。KEGG 通路注释是在evalue<0.000 01的条件下,利用blastx把unigene比对到KEGG数据库,然后根据比对结果查询与次级代谢产物合成通路相关的unigene。
1.3 SSR位点搜索和分析
利用MISA软件在所有unigene 中搜索SSR位点,参数设置如下:二核苷酸至少重复次数为6,三核苷酸、四核苷酸、五核苷酸和六核苷酸至少重复次数均为4[16]。
2 结果与分析
2.1 测序结果评估和读序列(reads)拼接组装
采用Illumina Hiseq2000测序平台,对女贞果实mRNA逆转录构建的cDNA文库进行测序,共获得32 856 614条clean reads,且本次测序总的cDNA碱基数约为4 928 492 100 bp(4.93 Gb),故每条reads的平均长度为150 bp。Q20及GC含量百分比依次为97.94%和43.97%(见表1)。由以上结果表明,转录组测序数据量和质量都较高,为后续的组装提供了较好的原始数据。
利用Trinity软件按paired-end方法对reads进行拼接组装,获得229 286个重叠群(contig),平均长为322 bp,总长为73 933 206 bp,N50为475 bp(见表1),其中小于200 bp 的重叠群数量占59.32%,200~3000 bp以及3000 bp以上各占40.16%和0.52%,具体长度分布见图1。所得重叠群再次组装共获得163 092条unigene,平均长为665.68 bp,总长为108 567 136 bp(108.57 Mb),N50为1345 bp(见表1),其中100~500、500~2000、2000 bp以上的各占64.01%、28.67%、7.32%,具体长度分布见图2。
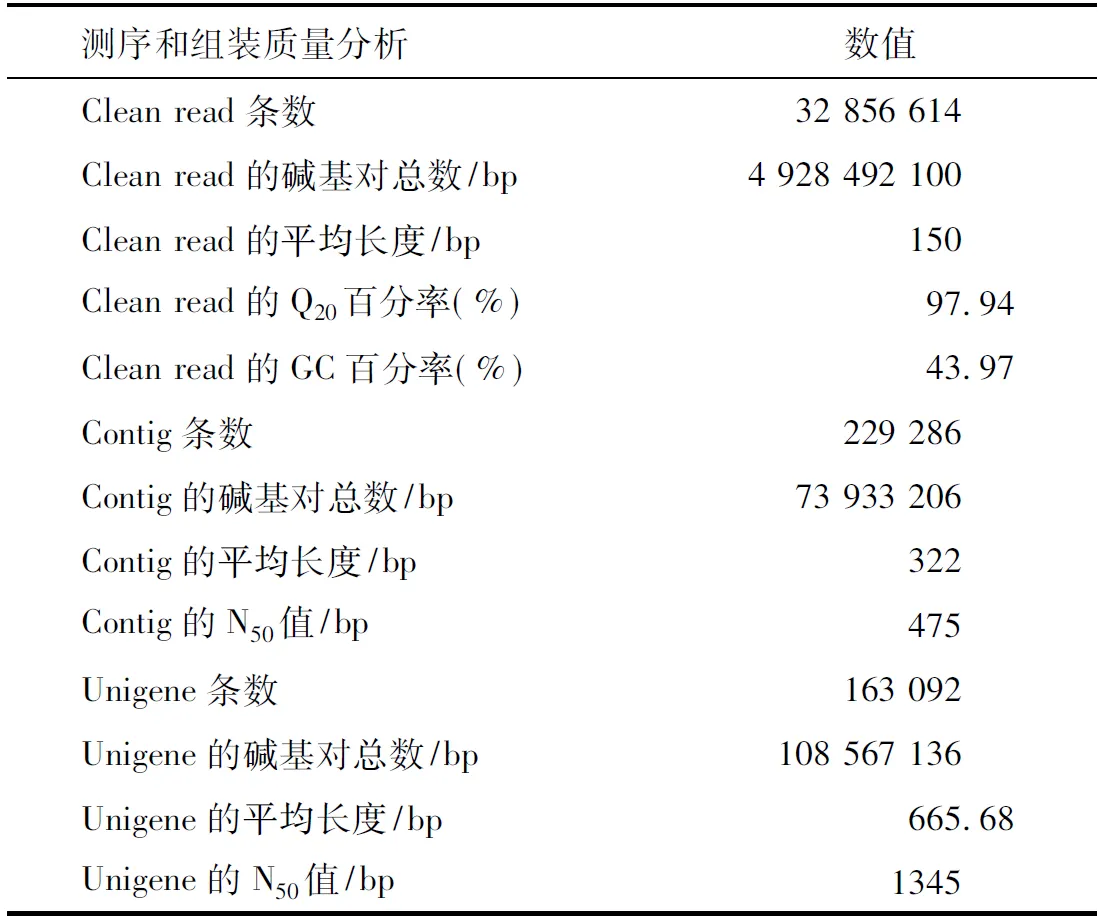
表1 女贞果实转录组测序和组装结果
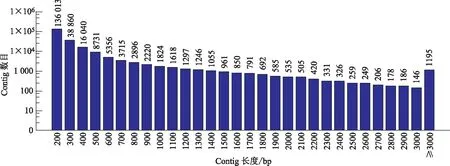
图1 Contig长度分布图
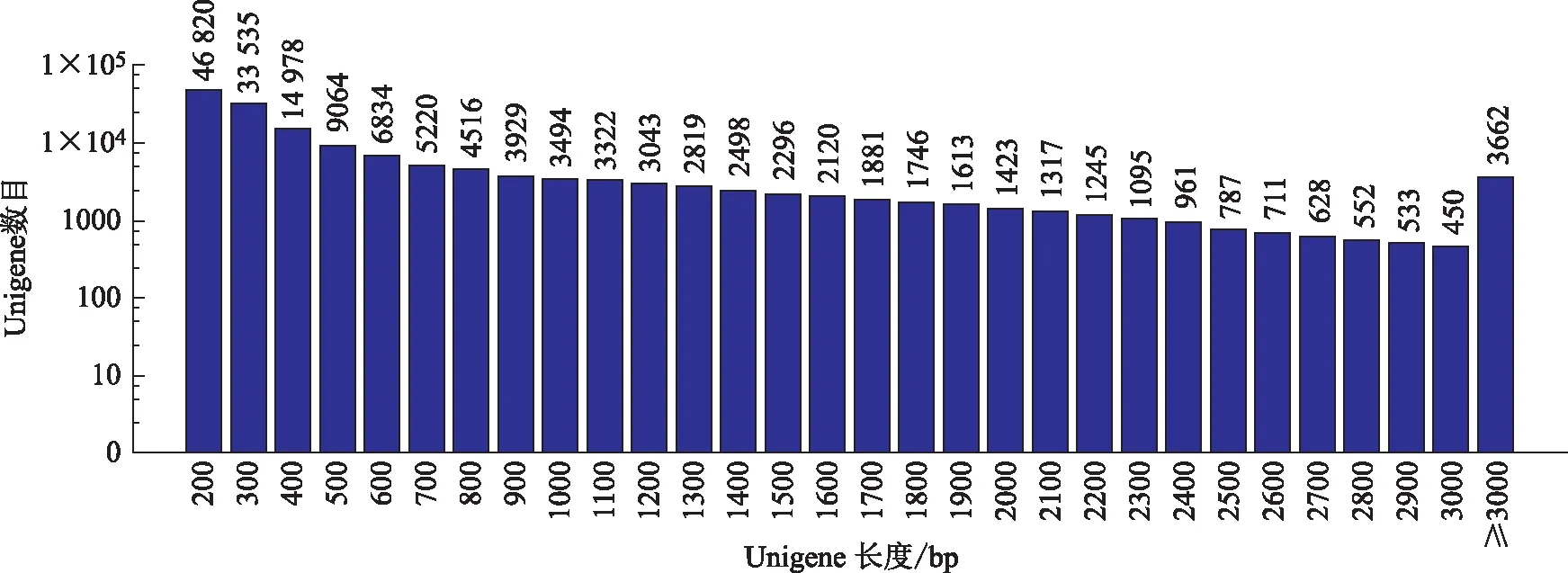
图2 Unigene长度分布图
2.2 序列比对和功能注释
将所获得的unigene与公共数据库Nr和Swiss-Prot进行Blastx比对,并利用Blastn比对Nt核酸数据库,结果显示共有83 903条unigene获得了基因注释,占unigene总数的51.45%。
2.3 功能分类和代谢途径分析
2.3.1 GO和COG功能分类 为进一步揭示女贞果实中unigene的功能分类,组装产生的163 092条unigene在GO(gene ontology)和COG(cluster of orthologous groups)数据库中分别进行比对分析。在GO分类中,共有16 876 条unigene被注释,其中12 203条unigene归入生物学过程类别,3015条unigene归入分子功能类别以及1658条unigene归入细胞组分类别(见图3)。在这3个类别中,生物学过程类别包括22个功能组,分子功能类别包括15个功能组,细胞组分类别则包括16个功能组(见图3)。代谢过程是生物学过程类别中最大功能组(9468条);细胞和细胞成分是细胞组分类别中最大功能组(都为5690条);催化活性是分子功能类别中最大功能组(9519条)。通过以上GO分析,使我们在转录水平对女贞果实功能基因的分类情况有了一个系统的了解。在COG分类中可将unigene分为25个类别(见图4),其中unigene条数最多的10个类别依次是一般功能预测类(6875条);复制、重组与修复类(3969条);转录类(3733条);信号转导机制类(2926条);蛋白质翻译后修饰、折叠和分子伴侣类(2632条);碳水化合物运输与代谢类(2198条);氨基酸运输与代谢类(1667条);功能未知类(1371条);细胞周期调控与分裂、染色体重排类(1201条)和无机离子运输与代谢类(1197条),而细胞外结构则是最小的类,仅包含4条unigene。通过以上COG分析,我们可以看到在女贞果实中,unigene涉及的功能类别主要是基因表达、蛋白质合成和物质代谢相关方面,这就正好符合果实作为一种营养器官,利用以上功能活动为自身积累营养物质这一基本功能。
2.3.2 KEGG代谢途径分析 KEGG(Kyoto encyclopedia of genes and genomes)可对细胞内unigene参与的代谢途径进行分析,本研究将获得的unigene比对到此数据库中,结果显示女贞果实的unigene主要参与了21类代谢途径(见图5),每条途径中又包括下一级具体的代谢过程,其中涉及主要的次级代谢产物黄酮类合成途径(Ko00941)含有258条unigene,萜类合成途径(Ko00900,Ko00902,Ko00904,Ko00909)含有929条unigene。以上这些unigene首次在女贞代谢途径中被发现,这为日后进一步验证它们的功能、并利用它们在体外生物合成相关的药用活性物质奠定了基础。
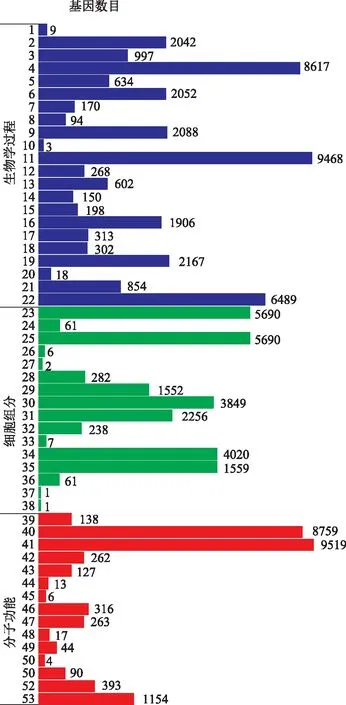
注:1.生物黏附;2.生物调节;3.细胞成分组织或生物发生;4.细胞进程;5.发育进程;6.定位的建立;7.生长;8.免疫系统进程;9.定位;10.运动;11.代谢进程;12.多有机体进程;13.多细胞有机体进程;14.生物进程的负调控;15.生物进程的正调控;16.生物进程调控;17.复制;18.生殖进程;19.刺激应答;20.节律进程;21.信号;22.单有机体进程;23.细胞;24.细胞连接;25.细胞成分;26.细胞外基质;27.细胞外基质成分;28.胞外区域;29.大分子复合物;30.膜;31.膜成分;32.膜关闭内腔;33.核区;34.细胞器;35.细胞器成分;36.共质体;37.病毒体;38.病毒体成分;39.抗氧化活性;40.结合;41.催化活性;42.电子载体活性;43.酶调节活性;44.鸟苷酸交换因子活性;45.金属伴侣活性;46.分子转导活性;47.核酸结合的转录因子活性;48.营养库活性;49.蛋白结合的转录因子活性;50.蛋白尾;51.受体活性;52.结构分子活性;53.转运活性。图3 女贞果实unigene的GO分类
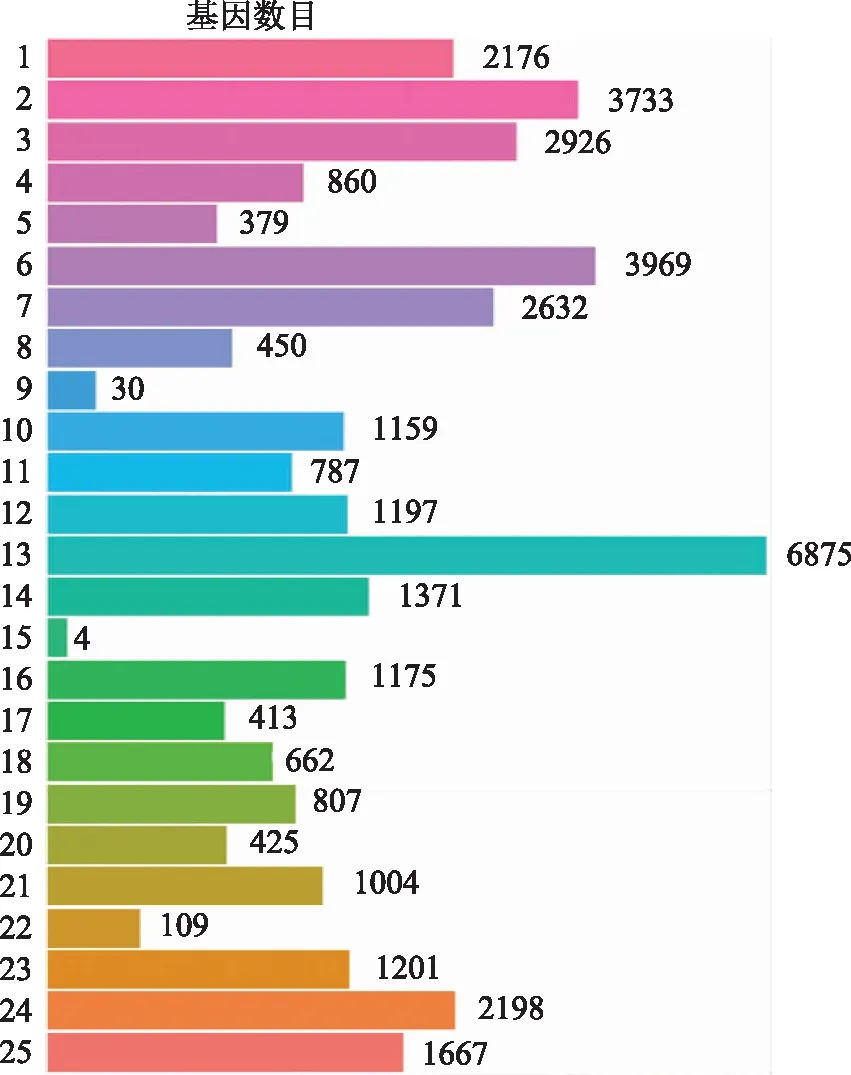
注:1.翻译,核糖体结构与生物发生;2.转录;3.信号传导机制;4.次级产物合成,运输及代谢;5.加工与修饰;6.复制、重组与修复;7.蛋白质翻译后修饰与转运,分子伴侣;8.核苷酸运输与代谢;9.核结构;10.脂类转运与代谢;11.细胞内转运,分泌和小泡运输;12.无机离子转运和代谢;13.一般功能预测;14.功能未知;15.胞外结构;16.能量生成和转换;17.防御机制;18.细胞骨架;19.辅酶运输和代谢;20.染色质结构与变化;21.细胞壁/膜生物发生;22.细胞运动;23.细胞周期控制,细胞分裂,染色体区分;24.碳水化合物运输和代谢;25.氨基酸运输与代谢。图4 女贞果实unigene的COG分类

注:1.运输与代谢;2.膜转运;3.信号转导;4.折叠,分类和降解;5.复制和修复;6.转录;7.翻译;8.药物抵抗;9.内分泌代谢病;10.氨基酸代谢;11.其他次生物质代谢;12.碳水化合物代谢;13.能量代谢;14.总览图;15.糖生物合成和代谢;16.脂质代谢;17.辅助因子和维生素代谢;18.其他的氨基酸代谢;19.萜类和聚酮类化合物的代谢;20.核苷酸代谢;21.环境适应。图5 女贞果实unigene的KEGG分类
2.4 SSR分析
利用SSR分析软件,从14 270条unigene中共搜索到15 925个SSR位点。搜索到的SSR位点类型丰富,单核苷酸至六核苷酸类型均有(见表2)。其中,二核苷酸重复所占比例最高,达到了43.95%;比例最低的是四核苷酸重复,仅为2.37%。在检测到的SSR中,出现频率最高的10类基序为:AG/GT(3856个)、AT/AT(183个)、AC/GT(125个)、AAT/ATT(940个)、AAG/CTT(736个)、ATC/ATG(501个)、ACC/GGT(491个)、AGC/CTG(316个)、AGG/CCT(312个)、CCG/CGG(133个)。

表2 女贞果实unigene中的SSR位点数量与分布
3 讨论
中药材是人类医药卫生领域的一个瑰宝,如何开发利用它使其不断的造福当代世人,一直是现今许多中医药科研工作者不断追求的目标。随着生物医药科研技术的不断发展,许多新技术新方法应用于中药材研究,使得以上目标逐步被实现,其中基于高通量测序技术的转录组学研究便是一个典型例子[17-18]。目前,转录组学在中药材研究领域主要集中于药用植物功能基因组的研究,即通过转录组学发现药用植物次生代谢产物生物合成关键酶基因、阐明次生代谢途径及其调控机制等,这将为利用功能基因和代谢途径体外生物合成中药材中药用活性成分,甚至是为创造性的研发新型药物奠定坚实的基础[19-20]。本研究正是基于以上描述,采用Illumina/Solexa高通量测序技术对女贞果实这一中药材进行了转录组测序,所得结果显示共有4.93 Gb有效数据产生,可组装拼接得到163 092个unigene,其中有79 189条unigene无法获得功能注释,究其原因:一是因为女贞缺乏基因组、EST和蛋白质序列信息,不能进行基因组条件下的比对分析,故导致较多的unigene无法匹配到合适的同源序列上;二是由于不能注释的unigene多为小于1000 bp的小片段,因而它们很难与公共数据库中已知的序列获得良好的比对;三是小片段的unigene有些可能是短的非编码序列或者新基因的某些不完整片段,从而导致它们无法被已有数据库所注释。另外,由测序数据组装拼接成unigene的过程中,N50值(指从组装最长的unigene依次向下求长度的总加和,当累加长度达到组装长度的一半时对应的unigene长度)是一个判断组装程度好坏的重要指标,其值越大说明组装得到的长片段越多,组装效果越好。本次研究unigene的N50值为1345 bp,大于1000 bp,说明本次序列组装的质量和长度可以满足转录组分析的基本要求,这为研究中进行以序列为基础的相关转录组分析提供了可靠保障。
转录组研究的一个重要目标是发现功能基因,并阐明功能基因编码产物所处的代谢途径。要实现这一目标,研究者需利用GO数据库、COG数据库和KEGG数据库对组装得到的unigene进行分析。本研究中笔者通过GO数据库的分析,首次系统地阐明了女贞果实功能基因的分类情况,并利用COG数据库对女贞果实unigene进行了功能注释,使其从分子水平找寻自己的直系同源体,从而预测它们的生物学功能,最后经KEGG数据库把一些预测的功能基因定位在了涉及主要次级代谢产物黄酮类和萜类合成的途径中。在此基础上,为进一步验证这些预测基因的真正功能,笔者将在后续的研究中对它们进行体外克隆和异源表达实验,并在体外和体内两个方面对表达产物进行功能活性检测,从而用实验结果检验转录组分析结果的正确与否。
SSR是真核生物基因组非编码区中存在的一些简单重复序列,在物种进化过程中呈现较为保守的特性,已广泛应用于中药材物种、种质资源以及农家种鉴定[21]。目前,女贞缺乏可使用的分子标记,此次果实转录组研究所获大量数据,可为开发女贞SSR标记提供丰富的筛选资源。在今后的研究中,可利用已获得的15 925个SSR位点,通过引物设计软件设计合理的SSR引物,然后利用SSR引物进行扩增检测,从中筛选出扩增稳定、条带清晰、多态性好的引物,为进一步开发女贞的SSR标记奠定基础[22]。
本研究利用Illumina/Solexa Hiseq2000测序平台首次对女贞果实进行了转录组测序研究,获得了大量的转录组数据。对这些数据从拼接组装、功能注释、代谢途径和SSR位点查询4个方面进行分析研究,使我们在转录水平对女贞果实有了比较详实的认识,同时也为今后开发利用女贞果实的药用次生代谢产物和分子标记物,以及为女贞基因组的测序组装提供了具有参考价值的基础数据。
[1] 国家药典委员会.中华人民共和国药典:一部[S].北京:中国医药科技出版社,2015:45-46.
[2] 尹辉.中药女贞子化学成分研究综述[J].九江学院学报(自然科学版),2015,30(1):74-75.
[3] 刘亭亭,王萌.女贞子化学成分与药理作用研究进展[J].中国实验方剂学杂志,2014,20(14):228-234.
[4] Nagalakshmi U,Waern K,Snyder M.RNA-Seq:A Method for Comprehensive Transcriptome Analysis[J].Curr Protoc Mol Biol,2010(suppl 89):Unit 4.11.1-13.
[5] Hoeijmakers W A,Bártfai R,Stunnenberg H G.Transcriptome analysis using RNA-Seq[J].Methods Mol Biol,2013,923:221-239.
[6] 田英芳,张晓政,周锦龙.转录组学研究进展及应用[J].中学生物教学,2013(12):29-31.
[7] Martin J A,Wang Z.Next-generation transcriptome assembly[J].Nature Reviews Genetics,2011,12(10):671-682.
[8] Chen C,Huang H,Wu C H.Protein Bioinformatics Databases and Resources[J].Methods Mol Biol,2011,694:3-39.
[9] Collins L J,Biggs P J,Voelckel C,et al.An approach to transcriptome analysis of non-model organisms using short-read sequences[J].Genome Informatics,2008,21:3-14.
[10] Martin M.Cutadapt removes adapter sequences from high-throughput sequencing reads[J].Embnet Journal,2011,17(1):10-12.
[11] Grabherr M G,Haas B J,Yassour M,et al.Full-length transcriptome assembly from RNA-Seq data without a reference genome[J].Nat Biotechnol,2011,29:644-652.
[12] Picardi E,Mignone F,Pesole G.EasyCluster:a fast and efficient gene-oriented clustering tool for large-scale transcriptome data[J].BMC Bioinformatics,2009,10(6):1-12.
[13] Bevilacqua V,Pietroleonardo N,Giannino E,et al.EasyCluster2:an improved tool for clustering and assembling long transcriptome reads[J].BMC Bioinformatics,2014,15(15):1-10.
[14] Conesa A,Götz S,García-Gómez J M,et al.Blast2GO:A universal tool for annotation,visualization and analysis in functional genomics research[J].Bioinformatics,2005,21:3674-3676.
[15] Ye J,Fang L,Zheng H,et al.WEGO:A web tool for plotting GO annotations[J].Nucleic Acids Res,2006,34:293-297.
[16] Wang H,Jiang J,Chen S,et al.Next-Generation Sequencing of the Chrysanthemum nankingense(Asteraceae)Transcriptome Permits Large-Scale Unigene Assembly and SSR Marker Discovery[J].PloS One,2013,8(4):e62293.
[17] 张晓萌,李健春,王琼,等.转录组测序技术在中医药领域的应用[J].中国现代中药,2016,18(8):1084-1087.
[18] 陈惠,辛丽丽,龚婕宁.基于全转录组测序技术的转录组学在中医药领域的应用前景分析[J].环球中医药,2013,6(10):759-763.
[19] 王尧龙,黄璐琦,袁媛,等.药用植物转录组研究进展[J].中国中药杂志,2015,40(11):2055-2061.
[20] 吴琼,孙超,陈士林,等.转录组学在药用植物研究中的应用[J].世界科学技术—中医药现代化,2010,12(3):457-462.
[21] 周骏辉,袁媛,黄璐琦.SSR标记在中药材分子身份证体系构建中的应用[J].中国现代中药,2016,18(10):1233-1236.
[22] 刘峰,王运生,田雪亮,等.辣椒转录组SSR挖掘及其多态性分析[J].园艺学报,2012,39(1):168-174.
