新型Sigma-1受体变构调节剂的发现和潜在应用
2018-06-19镇学初
王 允 镇学初
苏州大学药学院,苏州,215123,中国
Sigma-1受体的发现源于Martin教授1976年的报告。他们当时认为发现了一种新的阿片受体亚型,并将其命名其为“sigma opioids受体”,其特异性激动剂 为(+) SKF10047 及 pentazocine[1]。1982 年 Su 教授等利用放射自显影技术鉴定了一种与(+) SKF10047有nmol级亲和力的蛋白,然而该蛋白和阿片受体拮抗剂naloxone却并无亲和力。这就意味着可能该“(+)SKF10047结合的蛋白”不属于阿片受体。经过后续研究进一步证实后,Su教授重新命名其为sigma受体,以区别于阿片受体[2]。至今已知sigma受体存在两种亚型:sigma-1与sigma-2受体。Sigma-1受体已于1996年克隆,其晶体结构亦成功解析[3]。最新研究表明sigma-2受体即为跨膜蛋白97(transmembrane protein 97,TMEM97)蛋白[4]。本文主要关注 sigma-1 受体。
Sigma-1受体由223个氨基酸组成,主要位于内质网-线粒体接触面(endoplasmic reticulummitochondrion interface)的表面,也常被称作“线粒体相关的内质网膜(mitochondrion-associated endoplasmic reticulum membrane,MAM)”[2]。Sigma-1 受体由三个紧密相连的原体构成并具有两次跨膜结构(Fig.1)[3],分别由第8位到34位氨基酸、第83位到111位氨基酸组成,其N端和C端位于同一侧内质网腔隙。Sigma-1受体以寡聚体的形式存在(从六聚体到十五聚体不等),而激动剂常破坏其高聚状态。在不同物种中,sigma-1受体的配体结合域具有高度保守性[3];在哺乳动物中,sigma-1受体则具有超过90%的氨基酸同源性。在脑内,sigma-1受体在海马、丘脑等分布最为丰富,其次在纹状体、小脑、中缝背核、蓝斑核等有较丰富表达[3]。Sigma-1受体被认为是一种新型的伴侣蛋白,其伴侣活性可以被配基调控,具有受体的特点。正常情况下,位于MAM的免疫球蛋白结合蛋白(Binding immunoglobulin protein,Bip)结合sigma-1受体C端并抑制 sigma-1受体的活性;当sigma-1受体被激活后,表现为和Bip相分离,从MAM 转位到其他的区域发挥作用。
目前sigma-1受体的激活有两种形式:正位激动剂激活和变构调节剂激活。相对于广泛关注的sigma-1受体激动剂,其变构调节剂的研究较少但逐渐引起人们的重视,因此本文在综述sigma-1受体相关配基和生物学功能的基础上,重点介绍sigma-1受体变构调节剂的发现和潜在应用。
1 Sigma-1受体激动剂及拮抗剂

Fig.1 Structure of the sigma-1 receptor [3]
迄今为止,人们发现或合成了众多的sigma-1受体激动剂以及拮抗剂,比如常用激动剂有(+) pentazocine、(+) SKF10047、PRE-084 等(Tab.1),拮抗剂有 BD1047、BD1063等(Tab.2)[5]。事实上,众多神经精神类药物都和sigma-1受体有一定亲和力,如激动剂有成瘾类药物安非他明、可卡因,抗抑郁药物氟伏沙明、氟西汀,抗阿尔茨海默病药物多奈哌齐,拮抗剂有抗精神病药氟哌啶醇等等。大量研究证明这些药物的作用与其激活或拮抗 sigma-1 受体有关[6-7]。

Tab.1 Agonists of sigma-1 receptor[5]
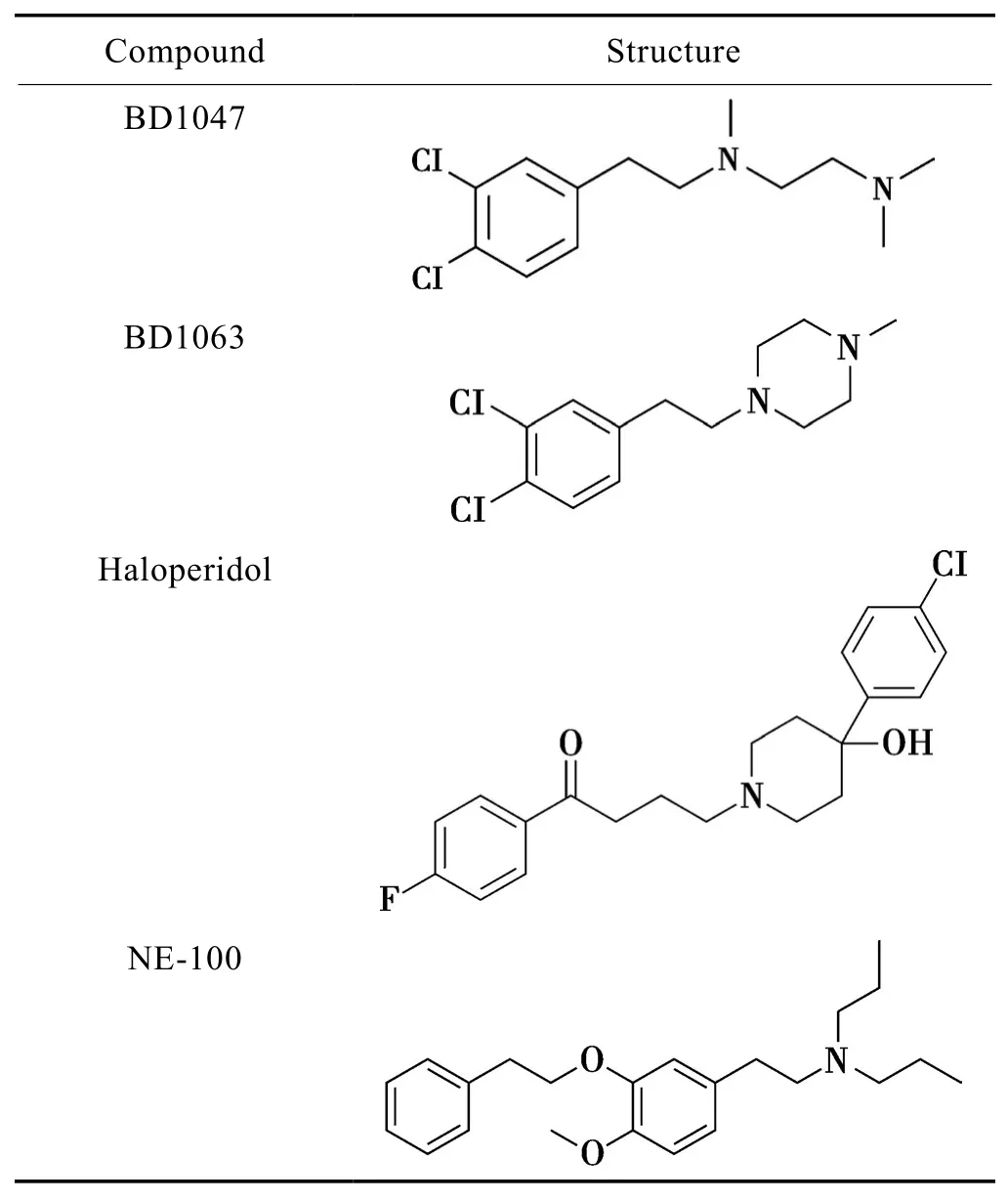
Tab.2 Antagonists of sigma-1 receptor[5]
一直以来人们都在寻找sigma-1受体真正的内源性配基。神经甾体类,比如黄体酮(progesterone),曾被认为可能是sigma-1受体的内源性配基[8]。生理条件下,progesterone可以达到400 nmol·L-1,但只有2%可以透过血脑屏障,以至脑内约只有8 nmol·L-1progesterone。加之有报道指出其与sigma-1受体的亲和力仍较低(Ki = 268或 36 nmol·L-1),因此多认为 progesterone 并非是 sigma-1受体内源性配基[9-10]。脱氢素雄酮(dehydroepiandrosterone,DHEA)在脑内含量丰富,有sigma-1受体激动作用,但亲和力低 (Ki = 706 nmol·L-1)[10-11]。二甲基色胺(N,N-dimethyltryptamine,DMT)曾被报道具有 sigma-1受体亲和力,由于其代谢快速,目前尚不能检测其脑内浓度。虽然DMT具有sigma-1受体激动剂的活性,比如可以抑制 INa,但是需要达到 100 μmol·L-1才能起效[12],加之其亲和力中等,同样人们也不认为其为sigma-1受体内源性激动剂。因此,到目前为止,sigma-1受体内源性配基尚未被真正公认。即便如此,以上的几个内源性物质仍被广泛用于sigma-1受体的功能研究中。
对于众多sigma-1受体配基而言,如何区分是激动剂还是拮抗剂曾一度困扰人们。随着sigma-1受体的分子生物学研究的突破,这一问题得到很好的解决。研究发现,sigma-1受体激动剂具备促进胞内sigma-1受体和Bip分离的作用,而拮抗剂对此没有影响但却能抑制激动剂活性。同时,激动sigma-1受体后也表现出相似的生物学效应。
2 Sigma-1受体生物学功能
2.1 调节离子通道活性
2.1.1 钾离子通道(potassium channels)
一直以来钾离子通道都被视为是sigma-1受体的重要作用靶点。Sigma-1受体可以通过直接或间接的作用抑制 K+通道。免疫共沉淀实验已经证实sigma-1受体可以分别和Kv1.2、Kv1.3、Kv1.4、hERG (human ether-a’-gogo K+channel)等 K+通道形成复合物[13-15]。Sigma-1受体激动剂如(+) pentazocine、JO-1784能够减少钾离子电流,抑制小电导钙离子激活的钾离子通道(small conductance calcium-activated K+channels,SK channels)[16-17]。
2.1.2 钙离子通道(calcium channels)
Sigma-1受体可以通过多种形式调节胞内钙离子浓度。其中,电压依赖的钙离子通道(voltage-dependent calcium channels,VDCCs)也是 sigma-1 受体发挥作用的靶点之一。激活sigma-1受体可以抑制L-型钙离子通道活性,比如激动剂(+) SKF-10047能明显抑制L-型钙通道电流[18]。也有报道指出,KCl诱导的NG108细胞去极化后,胞内钙离子浓度会增加,而激动剂 (+)pentazocine和PRE-084却对钙离子浓度发挥相反的调节作用;然而siRNA干扰sigma-1受体后它们的效应都能消失。因此,sigma-1受体对电压依赖的钙离子通道调控仍需进一步深入研究。
2.1.3 钠离子通道(sodium channels)
最初人们在海马脑片研究中发现,sigma-1受体激动剂硫酸脱氢表雄酮 (dehydroepiandrosterone sulphate,DHEAS)抑制一种持续钠电流(persistent Na+current,Na.p),且这种作用可能通过激活Gi蛋白-PKC信号通路[19]。而不同的实验研究则表明,sigma-1受体可以抑制Nav1.5通道电流,但并不需要ATP或GTP供能,提示sigma-1受体可能和Nav1.5通道存有直接作用[20]。Sigma-1受体激动剂1,3-di(2-tolyl)guanidine (DTG)、(+) pentazocine对钠离子通道的抑制作用会导致动作电位潜伏期延长,细胞放电率(fire rate)下降,同时使钠离子通道处于更稳定的灭活状态。
2.2 调控神经递质功能
Sigma-1受体参与调控许多神经递质系统释放,可以增加谷氨酸能、胆碱能、5-羟色胺能神经传递,减弱γ-氨基丁酸能、去甲肾上腺素能神经传递,调节多巴胺能神经传递。谷氨酸作为一类兴奋性神经递质在中枢神经系统分布最为广泛,研究表明,sigma-1受体可以增加谷氨酸能神经元的自发性放电,通过脑源性神经营养因子(brain-derived neurotrophic factor,BDNF)促进谷氨酸的释放,最终引起的胞内钙离子浓度升高。Sigma-1受体能与谷氨酸的N-甲基-D-天门冬氨酸(N-methyl-D-aspartate,NMDA)受体亚基GluN1形成复合物,同时激活的sigma-1受体还能增加与亚基GluN2A的相互作用,进而调控NMDA受体的功能[21]。
Sigma-1受体激动剂S-21377及S-21378能增加多巴胺能神经元的自发放电,同时可以增强NMDA受体诱导的中脑边缘及黑质纹状体通路多巴胺能神经元活性[22]。神经肽Y可以通过sigma-1受体增加海马胞外多巴胺的含量,而sigma-1受体又能增加金刚烷胺诱导的多巴胺能传递[23]。SA4503激动sigma-1受体后还可以增加多巴胺及其代谢产物DOPAC在前额叶皮质的含量[24]。
2.3 调控氧化应激、内质网应激和线粒体功能
氧的异常减少和自由基大量产生会导致细胞器、细胞或组织损伤。氧化应激也会对线粒体功能造成损伤,导致ATP大量消耗并降低抗氧化能力[25]。众多研究表明,SA4503、PRE-084等sigma-1受体激动可以明显降低氧化应激因素如H2O2造成的活性氧(reactive oxygen species,ROS)产生,而sigma-1受体基因被敲除或是表达被下调会增加细胞的氧化应激损伤[26-27]。体外实验研究证实,敲除sigma-1受体会导致内质网应激增加,反之,内质网应激增加能够上调sigma-1受体表达,而sigma-1受体过表达则有效抑制内质网应激损伤[28-29]。
Sigma-1受体和线粒体功能关系密切,转位至线粒体的sigma-1受体能和Rac1(一种小Rho家族GTP酶)形成复合物,进而调控线粒体活性氧的产生[30-31]。现已证实,sigma-1受体可以调控线粒体钙离子浓度。当内质网向胞内释放钙离子从而导致胞内钙离子浓度增加时,sigma-1受体可被激活,直接结合并稳定同样位于 MAM 的肌醇 1,4,5-三磷酸受体 3(inositol 1,4,5-triphosphate receptor type 3,IP3R-3),促进线粒体钙离子内流,从而影响线粒体功能。一项研究同样证实,激活sigma-1受体可以改善病理状态下心肌细胞线粒体钙离子浓度和ATP产生的减少[32]。
2.4 神经保护作用
Sigma-1受体对细胞生存有重要的意义。在神经元中,sigma-1受体激动剂可以稳定抗凋亡蛋白B淋巴细胞瘤 -2(B-cell lymphoma-2,Bcl-2),抑制促凋亡蛋白Bcl-2相关 X 蛋白(Bcl-2 associated X protein,Bax),并阻断caspase-3的激活[33-34]。虽然谷氨酸调控包括学习记忆在内的众多神经系统功能,然而谷氨酸的过度释放会造成NMDA受体持续激活,增加细胞内钙水平,导致钙稳态失衡引起细胞死亡。在体外缺血模型中,DTG、(+) pentazocine、PRE-084等激动 sigma-1受体显著降低谷氨酸的释放并阻断细胞内钙离子超载[35]。缺血后给予DTG 24 h能够有效减少梗死体积。选择性sigma-1受体激动剂SA4503能够显著缓解大鼠大脑中动脉闭塞模型中缺血梗死和行为学功能下降,表现出良好的应用前景[36]。
2.5 调控胶质细胞的活动
Sigma-1受体参与改善胶质细胞增生。最近研究发现,在肌萎缩侧索硬化症模型中,sigma-1受体激动剂PRE-084激活sigma-1受体后,抑制星形胶质细胞GFAP的表达[37]。Sigma-1受体也可以通过调控JAK2/STAT3信号通路抑制星形胶质细胞的激活[2]。在星形胶质细胞中,sigma-1受体激动剂BHDP能够保护缺氧诱导的ATP合成减少[38]。同样,在小胶质细胞模型中,sigma-1受体激动剂可以抑制小胶质的激活,显著抑制肿瘤坏死因子 -α(tumor necrosis factor-α,TNF-α)、诱导型一氧化氮合酶(inducible nitric oxide synthase,iNOS)、白细胞介素 -1β(interleukin-1β,IL-1β)等神经炎症因子的释放或表达[5-6]。
如上所述,sigma-1受体功能广泛而复杂。不仅对离子通道有重要调节作用,而且还对多种神经递质具有调控作用。这种作用不仅在神经元中,也体现在胶质细胞中。由于这种功能复杂性和该受体在中枢神经系统中分布的广泛性,以sigma-1受体激动剂为方向的药物,潜在的副作用问题可能比较突出。
3 Sigma-1受体变构调节剂
3.1 变构调节剂的优势
在受体结合位点上,变构调节剂(allosteric modulators)区别于正位激动剂(orthosteric agonists),一般可以分为以下三类:增强剂即正向变构调节剂(positive allosteric modulators,PAMs)、变构拮抗剂即负向变构调节剂(negative allosteric modulators,NAMs)和沉默变构调节剂(silent allosteric modulators,SAMs)[39]。 其 中,PAMs较为多见(本文主要涉及PAMs)。PAMs本身不具有类似激动剂的活性,但是可以通过稳定构象等方式提高正位激动剂和受体的亲和性,或者是降低了受体激活的能量障碍,或者两者同时作用。随着PAMs浓度的增加,量效曲线左移和上移的程度是有限的,当变构位点达到饱和时移动达到最大值。NAMs同样不具有内在活性,但结合变构调节位点后可以抑制正位激动剂的亲和力或者活性;SAMs能结合到变构位点但是不影响受体的功能。由于结合位点一样,SAMs的位点竞争会对别的变构调节剂的浓度应答曲线产生右移,就像在中性的正性拮抗剂对天然激活剂的量效曲线产生右移一样。
相对于传统的激动剂和拮抗剂等药物,理论上变构调节剂具有提高药物安全性和降低副作用等巨大的优势[40]。首先,变构调节剂具有更高的“时空可控性”。只有在内源性配基存在的情况下,变构调节剂才能发挥作用。内源性配基的作用一般受到体内精准调控,这也赋予变构调节剂精准性。可以体现为当体内配基不存在时,变构调节剂作用随即消失,表现为“时间性”。不同组织的内源性配基分布可能不同,这就赋予其更高的组织选择性,表现为“空间性”。再者,以变构增强剂为例,其激活效应有限度(其最大调节幅度受协同因子α限制),而外源性正位激动剂容易产生过度激活,产生不良反应以及发生受体脱敏等。
迄今,已经上市或处于药物研发不同阶段的变构小分子药物有近50种,例如靶向钙感应性受体(calcium-sensing receptor,CASR)以治疗继发性甲状旁腺功能亢进的药物Cinacalcet[41],以及靶向趋化因子受体 5(CC chemokine receptor 5,CCR5)以治疗艾滋病的药物 Maraviroc[42]等。
3.2 新型Sigma-1受体变构调节剂的发现
苯妥英(Phenytoin)是第一个被发现的sigma-1受体变构调节剂。Phentyoin能够增强sigma-1受体与多种激动剂((+) pentazocine,(+) SKF10047等)的结合[43-44],间接激活sigma-1受体。但其效能太低:体外实验显示phenytoin在10 μmol·L-1时才开始出现变构调节剂效应,1 mmol·L-1时达到其最大效应[45],因此无临床应用价值。
3.2.1 SKF83959
近年来,在研究单胺类递质受体过程中,发现了苯并氮杂类化合物 6-chloro-2,3,4,5-tetrahydro-3-methyl-1-(3-methylphenyl)-1H-3-benzazepine-7,8-diol(SKF83959)是一种新型sigma-1受体变构调节剂(Fig.2)[45]。在脑组织及肝脏中,SKF835959 能够促进3H( +) pentazocine(sigma-1受体激动剂)与sigma-1受体的结合,但不能促进3H-progesterone(sigma-1受体拮抗剂)与sigma-1受体的结合。SKF83959还能够促使sigma-1受体饱和曲线左移,降低sigma-1受体/3H( +)pentazocine复合物的解离速率。
同时,在肝脏但不是脑组织中,其结构类似物SCH22390、SKF38393也表现出相似的变构调节sigma-1受体作用。

Fig.2 The structure of SKF83959
3.2.2 SOMCL-668
尽管证实SKF83959是新型sigma-1受体变构调节剂(Fig.3),然而它并不是高选择性的sigma-1受体配基,它还是非典型多巴胺D1受体激动剂,并且与多种离子通道发生作用[46-50]。为了寻找更好选择性的sigma-1受体变构调节剂,我们在SKF83959的基础上进行了系列化学改造,最终发现3-methyl-phenyl-2,3,4,5-tetrahydro-1H-benzo[d]azepin-7-ol(SOMCL-668)具有更优的活性与选择性[51]。竞争结合实验表明它能增强激动剂(+) pentazocine与sigma-1受体的结合,不能增强拮抗剂progesterone与sigma-1受体的结合;而且 SOMCL-668 在 0.1 μmol·L-1时即能激活 sigma-1受体,10 μmol·L-1即可达到最大效应,其效能远远大于 phenytoin[45];除此以外,SOMCL-668 既不与多巴胺受体 D1、D2、5-羟色胺受体 5-HT1A、5-HT2A、去甲肾上腺素受体α1、α2等受体结合,也不直接影响Na+、Ca2+、K+、GABAA受体等离子通道活性[52-53]。

Fig.3 The structure of SOMCL-668
在功能上,SOMCL-668本身不能改变sigma-1受体和Bip的共定位或结合,但是可以明显促进激动剂诱导的sigma-1受体与Bip的分离;同时,SOMCL-668单独不能,但显著增加sigma-1受体刺激的sigma-1受体向细胞膜转位;此外SOMCL-668自身不具备,但是可以显著增加激动剂诱导的神经突起生长和BDNF分泌的能力。因此,SOMCL-668是国际上首个选择性sigma-1受体变构调节剂。
4 Sigma-1受体变构调节剂的潜在应用
4.1 抗癫痫作用
癫痫是大脑神经元突发性异常放电,导致短暂的大脑功能障碍的一种慢性疾病。虽然许多抗癫痫药被应用于临床治疗之中,但是这些药物仍不能满足临床需求。近来的研究显示某些sigma-1受体激动剂具有抗癫痫效应[54]。因此,以SKF83959为工具研究变构调节sigma-1受体是否具有抗癫痫作用。研究显示,SKF835959(20 和 40 mg·kg-1,ip)能有效拮抗戊四氮、电刺激和海人藻酸诱发的各种类型癫痫。其药理效应与sigma-1受体激动剂(+) SKF10047相当。而丙戊酸钠(valproicacid,VPA)则有效抑制电刺激诱发的强直性惊厥和戊四氮诱发的多种惊厥,但是不能改善海人藻酸所诱导的癫痫持续状态。这说明SKF83959具有广泛的抗癫痫效应。还发现sigma-1受体的拮抗剂BD1047能够逆转SKF83959的抗癫痫效应,而D1受体拮抗剂SCH23390则不影响SKF83959的抗癫痫效应;从而提示SKF83959的抗癫痫效应依赖于sigma-1受体的活化[55]。
以VPA为代表的经典抗癫痫药往往会具有镇静、运动功能受损等不良反应。也发现,sigma-1受体激动剂(+) SKF10047在治疗效量时也会影响动物的协调能力。不同于 VPA 和(+) SKF10047,sigma-1受体变构调节剂SKF83959并不影响动物的自发活动和运动协调能力。这提示sigma-1受体变构调节剂用于癫痫治疗时,其不良反应少于传统的抗癫痫药物[56]。
这一发现首次证明sigma-1受体变构调节剂SKF83959显示出广泛的抗癫痫活性,为sigma-1受体别构调节剂用于癫痫治疗提供了实验证据。
4.2 抑制神经炎症作用
在帕金森病等神经退行性病变的发生与发展过程中,脑内始终存在以胶质细胞,特别是小胶质细胞激活为主要特征的炎症反应[57]。小胶质细胞作为中枢神经系统常驻固有免疫巨噬细胞,维持中枢神经系统正常的内环境稳态,扮演着支持、营养、信号传导、保护、修复等重要角色。然而小胶质细胞过度激活会引起大量的促炎细胞因子释放,从而诱导神经炎症发生,造成神经元的损伤,进而促进神经退行性病变的发生。
本实验室早期的一项动物实验证实SKF83959能够改善帕金森病的作用[58],因此我们重点研究了SKF83959作为sigma-1受体变构调节剂对神经炎症的作用。在脂多糖(lipopolysaccharide,LPS)介导的BV2小胶质细胞持续激活状态下,SKF83959 显著抑制LPS激活的BV2 小胶质细胞炎症反应,但可被sigma-1受体拮抗剂BD1047和BD1063以及DHEA合成抑制剂酮康唑阻断;SKF83959促进了DHEA与sigma-1受体结合,并以正向协同的方式增强了外源性DHEA的抗炎作用;sigma-1受体拮抗剂 BD1047和BD1063也阻断了SKF83959和DHEA的协同抗炎作用;在小胶质细胞条件培养实验中,SKF83959可以改善小胶质细胞炎症介质对HT-22神经元的神经毒性;SKF83959的抗炎作用与 MAPK/ERK和 NF-κB信号通路激活以及多巴胺D1受体活化并不相关[59]。这揭示了SKF83959作为sigma-1受体变构调节剂的抗炎作用新机制,也提示sigma-1受体变构调节剂可能在改善神经炎症中存有潜在应用[59]。
4.3 抗抑郁作用
抑郁症是一种最为常见的情感类疾病,严重危害人类健康[60]。以氟西汀、文拉法辛等为代表的选择性单胺类递质再摄取抑制剂存在治愈率低、起效慢等缺点[61]。因此,寻求新的治疗靶点尤其是起效时间优于现有治疗靶点的抗抑郁药物具有重大意义。
本实验室采用小鼠强迫游泳、悬尾实验、慢性不可预知性温和刺激等评价sigma-1受体变构调节剂SOMCL-668的抗抑郁作用。结果显示,SOMCL-668可以减少强迫游泳、悬尾实验中小鼠不动时间,表现出抗抑郁作用,提前给予sigma-1受体拮抗剂BD1047则取消其抗抑郁作用;SOMCL-668可以改善抑郁症模型中小鼠快感缺乏、焦虑及体质量下降等症状,同时可以缓解抑郁症导致的BDNF及糖原合成酶激酶 -3β(glycogen synthase kinase-3β,GSK-3β) 磷 酸化水平(ser-9)的下调,并且SOMCL-668抗抑郁起效时间优于现有抗抑郁药物文拉法辛。在动物实验中,SOMCL-668可以快速增加小鼠海马中GSK3β磷酸化水平(ser-9),sigma-1受体拮抗剂BD1047则阻断这种作用;在细胞实验中,SOMCL-668可以通过变构调节的方式增加HT-22细胞中GSK3β磷酸化水平(ser-9),siRNA沉默sigma-1受体表达后同样取消这种作用。药理性激活GSK3β可以阻断SOMCL-668在强迫游泳及悬尾中的抗抑郁作用。这说明sigma-1受体变构调节SOMCL-668具有快速抗抑郁作用,其抗抑郁作用可能和 BDNF-GSK3β通路有关[41]。
4.4 其他
国外有课题组最近也报道了另外一种新型sigma-1受体变构调节剂(4R,5S)-2-(5-methyl-2-oxo-4-phenylpyrrolidin-1-yl)-acetamide (methylphenylpiracetam,E1R)(Fig.4)。被动逃避实验显示,该化合物可以增强小鼠认知功能;被动逃避及Y迷宫实验显示,该化合物可以缓解东莨菪碱所致的小鼠认知功能损伤[62]。同时,另一项体外实验显示sigma-1受体变构调节剂还具有促进神经嵴干细胞向施旺细胞分化的作用[63]。

Fig.4 The structure of E1R
5 结语
目前,已公开报道的sigma-1受体变构调节剂数量较少。这和人们对sigma-1受体变构调节方式研究不足有关。人们并不清楚变构调节的具体作用位点,偶然获取仍是sigma-1受体变构调节剂的主要来源,这大大限制了对变构调节剂的主动发现。其次,变构调节剂的优势仍主要体现在理论上,尚需要深入和系统研究以证实其优越性。
总体而言,作为一种直接快速和广泛有效的蛋白功能调节方式,变构调节位点具有高度多样性,可以成为多个小分子的结合位点;别构调节小分子不结合受体正位活性位点上,不直接影响受体与其激动剂的正常结合,其对受体调节效应有一定上限,不良反应小。因此以上优点使得sigma-1受体变构调节剂成为药物发现的热门方向。
[1] Ajmo C T,Vernon D O,Collier L,et al. Sigma receptor activation reduces infarct size at 24 hours after permanent middle cerebral artery occlusion in rats[J].Current Neurovascular Research,2006,3: 89-98.
[2] Matthew J Robson,Ryan C Turner,Zachary J Naser,et al. SN79,a sigma receptor antagonist,attenuates methamphetamine-induced astrogliosis through a blockade of OSMR/gp130 signaling and STAT3 phosphorylation[J].Experimental neurology,2014,254:180-189.
[3] Schmidt H R,Zheng S,Gurpinar E,et al. Crystal structure of the human sigma1 receptor[J].Nature,2016,532: 527-530.
[4] Assaf Alon,Hayden R Schmidt,Michael D Wood,et al.Identification of the gene that codes for the sigma 2 receptor[J].Proceedings of the National Academy of Sciences of the United States of America,2017,114(27): 7160-7165.
[5] Cobos E J,Entrena J M,Nieto F R,et al. Pharmacology and therapeutic potential of sigma(1) receptor ligands[J].Current Neuropharmacology,2008,6: 344-366.
[6] Zhao Jing,Ha Yon-ju,Gregory I Liou,et al. Sigma receptor ligand,(+) -pentazocine,suppresses in flammatory responses of retinal microglia[J].Invest Ophthalmol Vis Sci,2014,55(6): 3375-3384.
[7] Hall A A,Herrera Y,Ajmo C T,et al. Sigma receptors suppress multiple aspects of microglial activation[J].Glia,2009,57: 744-754.
[8] Mollyy Johannessen,Dominique Fontanilla,Timur Mavlyutov,et al. Antagonist action of progesterone at sigmareceptors in the modulation of voltage-gated sodium channels[J].Am J Physiol Cell Physiol,2011,300:C328-C337.
[9] Schwarz S,Pohl P,and Zhou G Z. Steroid binding at sigma-“opioid” receptors[J].Science,1989,246: 1635-1638.
[10] Kenji Hashimoto. Activation of sigma-1 receptor chaperone in the treatment of neuropsychiatric diseases and its clinical implication[J]. J pharmacological Sciences,2015,127(1):6-9.
[11] Rikki Waterhouse,Raymond C Chang,Nana Atuehene,et al. In vitro and in vivo binding of neuroactive steroids to the sigma-1 receptor as measured with the positron emission tomography radioligand [18F]FPS[J].Synapse,2007,61:540-546.
[12] Dominique Fontanilla,Molly Johannessen,Abdol R Hajipour,et al. The hallucinogen N,N-dimethyltryptamine(DMT) is an endogenous sigma-1 receptor regulator[J].Science,2009,323(5916):934-937.
[13] Maho Kinoshita,Yoshikazu Matsuoka,Takeshi Suzuki,et al.Sigma-1 receptor alters the kinetics of Kv1.3 voltage gated potassium channels but not the sensitivity to receptor ligands[J].Brain Res,2012,1452: 1-9.
[14] Dilshan Balasuriya,Lauren D’Sa,Ronel Talker,et al. A direct interaction between the sigma-1 receptor and the hERG voltage-gated K+ channel revealed by atomic force microscopy and homogeneous time-resolved fluorescence (HTRF(R)) [J].J Biol Chem,2014,289: 32353-32363.
[15] Said Kourrich,Teruo Hayashi,Jian-ying Chuang,et al.Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine[J].Cell,2013,152(1-2): 236-247.
[16] Marzia Martina,Turcotte M.E.,Halman S.,et al. The sigma-1 receptor modulates NMDA receptor synaptic transmission and plasticity via SK channels in rat hippocampus[J]. J Physiology,2007,578: 143-157.
[17] Olivier Soriani,Frank Le Foll,Francois Roman,et al.A-Current down-modulated by sigma receptor in frog pituitary melanotrope cells through a G protein-dependent pathway[J].J Pharmacol Exp Ther,1999,289(1): 321-328.
[18] Tchedre K T,Huang R Q,Dibas A,et al. Sigma-1 receptor regulation of voltage-gated calcium channels involves a direct interaction[J].Invest Ophthalmol Vis Sci,2008,49(11):4993-5002.
[19] Cheng Zheng-xiang,Lan Dan-mei,Wu Pei-ying,et al.Neurosteroid dehydroepiandrosterone sulphate inhibits persistent sodium currents in rat medial prefrontal cortex via activation of sigma-1 receptors[J].Experimental Neurology,2008,210(1): 128-136.
[20] Molly Johannessen,Subramaniam Ramachandran,Logan Riemer,et al. Voltage-gated sodium channel modulation by sigma-receptors in cardiac myocytes and heterologous systems[J].Am J Physiol Cell Physiol,2009,296: C1049-C1057.
[21] Mohan Pabba M,Adrian Y C Wong,Nina Ahlskog,et al. NMDA receptors are upregulated and trafficked to the plasma membrane after sigma-1 receptor activation in the rat hippocampus[J].J Neurosci,2014,34(34): 11325-11338.
[22] Benjamin Gronier,Guy Debonnel. Involvement of sigma receptors in the modulation of the glutamatergic/NMDA neurotransmission in the dopaminergic systems[J].Eur J Pharmacol,1999,368: 183-196.
[23] Magali Peeters,Pascal Romieu,Tangui Maurice,et al.Involvement of the sigma 1 receptor in the modulation of dopaminergic transmission by amantadine[J]. European J Neuroscience,2004,19: 2212-2220.
[24] Tetsuya Kobayashi,Kiyoshi Matsuno,Masaaki Murai,et al. Sigma 1 receptor subtype is involved in the facilitation of cortical dopaminergic transmission in the rat brain[J].Neurochem Res,1997,22(9): 1105-1109.
[25] Lin M T,Beal M F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases[J].Nature,2006,443:787-795.
[26]Arindam Pal,Dominique Fontanilla,Anupama Gopalakrishnan,et al. The sigma-1 receptor protects against cellular oxidative stress and activates antioxidant response elements[J].Eur J Pharmacol,2012,682(1-3): 12-20.
[27] Tuerhong Tuerxun,Tadahiro Numakawa,Naoki Adachi,et al. SA4503,a sigma-1 receptor agonist,prevents cultured cortical neurons from oxidative stress-induced cell death via suppression of MAPK pathway activation and glutamate receptor expression[J].Neurosci Lett,2010,469(3): 303-308.
[28]Tomohisa Mori,Teruo Hayashi,Eri Hayashi,et al. Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival[J].PLoS One,2013,8: e76941.
[29] Teruo Hayashi,Su Tsung-Ping. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival[J].Cell,2007,131(3): 596-610.
[30] Nino Natsvlishvili,Nino Goguadze,Elene Zhuravliova,et al.Sigma-1 receptor directly interacts with Rac1-GTPase in the brain mitochondria[J].BMC Biochemistry,2015,16: 11.
[31] Heather L Osborn-Heaford,Alan J Ryan,Shubha Murthy,et al. Mitochondrial Rac1 GTPase import and electron transfer from cytochrome c are required for pulmonary fibrosis[J].J Biol Chem,2012,287: 3301-3312.
[32] Hideaki Tagashira,Md Shenuarin Bhuiyan,Norifumi Shioda,et al. Fluvoxamine rescues mitochondrial Ca2+transport and ATP production through sigma(1)-receptor in hypertrophic cardiomyocytes[J].Life Sci,2014,95(2): 89-100.
[33] Kissaou T Tchedre,Thomas Yorio. Sigma-1 receptors protect RGC-5 cells from apoptosis by regulating intracellular calcium,Bax levels,and caspase-3 activation[J].Invest Ophthalmol Vis Sci,2008,49: 2577-2588.
[34] Yang Su-fang,Anish Bhardwaj,Cheng Jian,et al. Sigma receptor agonists provide neuroprotection in vitro by preserving Bcl-2[J].Anesthesia and Analgesia,2007,104:1179-1184.
[35] Katnik C,Guerrero W R,Pennypacker K R,et al. Sigma-1 receptor activation prevents intracellular calcium dysregulation in cortical neurons during in vitro ischemia[J].J Pharmacol Exp Ther,2006,319: 1355-1365.
[36] Karsten Ruscher,Mehrdad Shamloo,Mattias Rickhag,et al. The sigma-1 receptor enhances brain plasticity and functional recovery after experimental stroke[J].Brain,2011,134(3): 732-746.
[37] Marco Peviani,Eleonora Salvaneschi,Leonardo Bontempi,et al. Neuroprotective effects of the Sigma-1 receptor (S1R)agonist PRE-084,in a mouse model of motor neuron disease not linked to SOD1 mutation[J].Neurobiol Dis,2014,62:218-232.
[38] Anis Klouz,Jean-Paul Tillement,Marie-Francoise Boussard,et al. [3H]BHDP as a novel and selective ligand for sigma1 receptors in liver mitochondria and brain synaptosomes of the rat[J].FEBS Lett,2003,553(3): 157-162.
[39] Wang Li-yun,Martin Bronwen,Randall Brenneman,et al. Allosteric modulators of g protein-coupled receptors:future therapeutics for complex physiological disorders[J].J Pharmacol Exp Ther,2009,331(2): 340-348.
[40] Stephan Urwyler. Allosteric modulation of family C G-protein-coupled receptors: from molecular insights to therapeutic perspectives[J].Pharmacol Rev,2011,63(1):59-126.
[41] Alice Cavanaugh,Huang Ying,Gerda E Breitwieser.Behind the curtain: cellular mechanisms for allosteric modulation of calcium-sensing receptors[J].British Journal of Pharmacology,2012,165: 1670-1677.
[42] Bernard Lagane,Javier Garcia-Perez,Esther Kellenberger.Modeling the allosteric modulation of CCR5 function by Maraviroc[J].Drug Discovery Today Technologies,2013,10(2): e297-e305.
[43] Enrique J Cobos,Jose M Baeyens,Esperanza Del Pozo.Phenytoin differentially modulates the affinity of agonist and antagonist ligands for sigma 1 receptors of guinea pig brain[J].Synapse,2005,55: 192-195.
[44] Cobos E J,Lucena G,Baeyens J M,et al. Differences in the allosteric modulation by phenytoin of the binding properties of the sigma1 ligands [3H](+)-pentazocine and [3H]NE-100[J].Synapse,2006,59: 152-161.
[45] Guo Lin,Zhao Jiang-hao,Jin Guo-zhang,et al.SKF83959 is a potent allosteric modulator of sigma-1 receptor[J].Mol Pharmacol,2013,83: 577-586.
[46] Zhou Shang-lin,Chu Hong-yuan,Jin Guo-zhang,et al.Effects of SKF83959 on the excitability of hippocampal CA1 pyramidal neurons: a modeling study[J].Acta Pharmacol Sin,2014,35: 738-751.
[47] Fang Xing,Guo Lin,Xu Jia-Jia,et al. SKF83959 is a novel triple reuptake inhibitor that elicits anti-depressant activity[J].Acta Pharmacol Sin,2013,34: 1149-1155.
[48] Chu Hong-yuan,Wu Qian-qian,Zhou Shang-lin,et al.SKF83959 suppresses excitatory synaptic transmission in rat hippocampus via a dopamine receptor-independent mechanism[J].J Neurosci Res,2011,89: 1259-1266.
[49] Chu Hong-yuan,Gu Qin-hua,Jin Guo-zhang,et al.Electrophysiological effects of SKF83959 on hippocampal CA1 pyramidal neurons: potential mechanisms for the drug’s neuroprotective effects[J].PLoS One,2010,5(10):e13118.
[50] Chen Xue-qin,Zhang Jing,John L Neumeyer,et al.Arylbenzazepines are potent modulators for the delayed rectifier K+channel: a potential mechanism for their neuroprotective effects[J].PLoS One,2009,4: e5811.
[51] 镇学初,张翱,郭琳.苯并氮杂内化合物在制备预防或治疗癫痫药物中的应用[P].中国专利No. 201210337109,2013.
[52] Zhang Jing,Huang Ji-ye,Song Zi-lan,et al. Structural manipulation on the catecholic fragment of dopamine D(1)receptor agonist 1-phenyl-N-methyl-benzazepines[J].European J Medicinal Chemistry,2014,85: 16-26.
[53] Guo Lin,Chen Yan-ke,Zhao Rui,et al. Allosteric modulation of sigma-1 receptors elicits anti-seizure activities[J].British J Pharmacology,2015,172: 4052-4065.
[54] Edijs Vavers,Baiba Svalbe,Lasma Lauberte,et al. The activity of selective sigma-1 receptor ligands in seizure models in vivo[J].Behavioural Brain Research,2017,328: 13-18.
[55] Anders A Jensen,Hans Brauner-Osborne. Allosteric modulation of the calcium-sensing receptor[J].Current Neuropharmacology,2007,5(3): 180-186.
[56] Marcia M Moraes,Marcella C Galvao,Danilo Cabral,et al. Propentofylline prevents sickness behavior and depressivelike behavior induced by lipopolysaccharide in rats via neuroinflammatory pathway[J].PLoS One,2017,12:e0169446.
[57] Michael T Heneka,Markus P Kummer,Eicke Latz. Innate immune activation in neurodegenerative disease[J].Nature Reviews Immunology,2014,14: 463-477.
[58] Zhang Hai,Ma Li-qun,Wang Fang,et al. Chronic SKF83959 induced less severe dyskinesia and attenuated L-DOPA-induced dyskinesia in 6-OHDA-lesioned rat model of Parkinson’s disease[J].Neuropharmacology,2007,53: 125-133.
[59] Christopher Bown,Wang Jun-feng,Glenda MacQueen,et al. Increased temporal cortex ER stress proteins in depressed subjects who died by suicide[J].Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology,2000,22: 327-332.
[60] Aliza J Ferrari,Fiona J Charlson,Rosana Elizabeth Norman,et al. The epidemiological modelling of major depressive disorder: application for the Global Burden of Disease Study 2010[J].PLoS One,2013,8: e69637.
[61] Andre Tadic,Stefanie Wagner,Stanislav Gorbulev,et al.Peripheral blood and neuropsychological markers for the onset of action of antidepressant drugs in patients with Major Depressive Disorder[J].BMC Psychiatry,2011,11: 16.
[62] Matthew A Timberlake,Yogesh Dwivedi. Altered expression of endoplasmic reticulum stress associated genes in hippocampus of learned helpless rats: relevance to depression pathophysiology[J].Frontiers in Pharmacology,2015,6:319.
[63] Philip Gold,Julio Licinio,Maria G Pavlatou. Pathological parainflammation and endoplasmic reticulum stress in depression: potential translational targets through the CNS insulin,klotho and PPAR-gamma systems[J].Mol Psychiatry,2013,18: 154-165.
