谷氨酸功能异常与阿尔茨海默病
2018-06-17张帅艾静
张 帅 艾 静
哈尔滨医科大学药学院药理教研室,哈尔滨,150086,中国
谷氨酸是中枢神经系统内重要的兴奋性神经递质,由突触前神经元合成和储存,在神经冲动刺激下释放入突触间隙,与突触后膜上的谷氨酸受体结合并发挥功能[1]。谷氨酸作为神经递质,主要参与神经突触可塑性形成及学习与记忆的形成过程[2-3]。
阿尔茨海默病(Alzheimer's disease,AD)是一种中枢神经系统变性病,起病隐匿,病程呈慢性进行性,是老年期痴呆中最常见的一种类型[4-5]。AD的临床特征是记忆力进行性减退,认知功能障碍,行为活动异常等[4],其病理学特征是在患者大脑皮质,海马和某些皮质下神经核团形成老年斑,神经元纤维缠结,颗粒空泡变性和Hirano小体[6]。由于AD的发病机制尚不十分清楚,目前没有有效的预防和根治方法。细胞外谷氨酸浓度的改变与认知障碍密切相关,谷氨酸介导的神经突触传递过程在AD中也显著受累[7-8]。因此,本文主要论述谷氨酸功能异常在AD中的作用,为AD治疗药物靶点的发现和新药研究提供借鉴。
1 谷氨酸合成,释放及重吸收功能与AD
1.1 谷氨酸合成,释放及重吸收的生理过程
动物体内谷氨酸的来源有两种,一种源于葡萄糖代谢:葡萄糖经无氧糖酵解生成丙酮酸,之后丙酮酸进入线粒体经氧化脱羧生成乙酰辅酶A并进入三羧酸循环,经一系列步骤合成α-酮戊二酸,α-酮戊二酸在谷氨酸脱氢酶的作用下产生谷氨酸,这类谷氨酸被认为主要起代谢作用,与蛋白质的合成等相关;第二种来源的谷氨酸则主要作为兴奋性神经递质存在,通过谷氨酸-谷氨酰胺循环由谷氨酰胺通过谷氨酰胺酶催化脱氨基形成[9],谷氨酰胺酶由于可以被磷酸盐刺激激活而被称为磷酸活化谷氨酰胺酶(phosphate activated glutaminase,PAG),催化谷氨酰胺水解生成谷氨酸和氨[10]。合成的谷氨酸被转运蛋白-囊泡谷氨酸转运体(vesicular glutamate transporters,VGLUTs)以ATP依赖的方式摄取并浓缩于突触小泡中[11]。成熟的突触前末端由三个突触小泡池组成,通常称为预备释放池(readily releasable pool,RRP),循环池(recycling pool)和储存池(reserve pool,RP)。RRP中囊泡在受到刺激后立即可用,RP则被定义为突触小泡的储存室(占全部小泡80%~90%),突触小泡从这里的释放需要由强烈刺激引发,循环池小泡占全部小泡的5%~20%,以适度刺激可释放[12]。
谷氨酸从突触前释放至突触间隙是一个钙离子依赖性的过程[13],当突触前神经元兴奋时,动作电位沿轴突传递到突触前膜使突触前膜发生去极化,引起突触前膜电压依赖性Ca2+通道开放,Ca2+内流,Ca2+与钙调蛋白结合,激活钙调蛋白依赖性蛋白激酶Ⅱ(calmodulin-dependent protein kinaseⅡ,CaMKⅡ),使突触蛋白Ⅰ(synapsinⅠ)磷酸化,突触囊泡从细胞骨架上游离出来。存在于突触囊泡膜上的小突触泡蛋白(synaptobriven)与存在于突触前膜上的突触融合蛋白(syntaxin)和SNAP-25合称为SNARE复合物,SNARE复合物向活化区募集Ca2+通道,然后经Rab3A蛋白促进突触小泡与突触前膜融合[14-15]。SNARE复合物的装配也会促进Ca2+的内流并增加融合孔的开放率,此外,脑源性神经营养因子(brain-derived neurotrophic factor,BDNF)可以通过TrkB/Src/PLC-γ1通路促进synapsinⅠ和β-连环蛋白(β-catenin)磷酸化来促进突触小泡锚定并增加谷氨酸的量子化释放[16-18]。
发挥作用后,突触间隙的谷氨酸会被迅速去除,以防止兴奋性毒性的发生[19]。谷氨酸重吸收的方式有两种:一种是被突触前末端再摄取,另一种是在突触间隙扩散并被胶质细胞摄取[20],后者主要依赖于星形胶质细胞上兴奋性氨基酸转运蛋白(excitatory amino acid transporters,EAATs)的转运[21-22],被星形胶质细胞摄取的谷氨酸转变为谷氨酰胺并被运输到细胞外间隙,谷氨酰胺被神经元摄取并重新转化为谷氨酸[23],然后经VGLUT浓缩到突触前膜的囊泡中等待释放[24](Fig.1)。
1.2 谷氨酸合成,释放及重吸收功能异常与AD
从正常认知发展到认知功能障碍甚至AD的疾病进程中,中枢神经系统中谷氨酸的水平会呈现波动变化。研究表明谷氨酸功能异常的出现可能先于认知障碍的发生[25-26]。通过对APP/PS1小鼠的研究发现,在未出现显著认知功能障碍之前(2~4月),小鼠海马CA1区谷氨酸功能便出现异常[25]。虽然2~4月龄APP/PS1小鼠脑中谷氨酸基础水平未出现改变,但钾离子诱导的谷氨酸释放显著增加,谷氨酸回收率也明显增加,胶质细胞功能标记物肌醇(myo-inositol,mIn)上调,这些改变被认为是脑内的一种代偿性机制[26],而CA3及DG区未出现此种改变。随着年龄的增加,5~8月APP/PS1小鼠脑内谷氨酸基础水平已经出现明显的年龄依赖性下降,钾离子诱发谷氨酸释放水平的升高与小鼠空间记忆呈负相关[26]。在体及海马脑片研究发现,低浓度(0.01及0.10 μmol·L-1)的Aβ可以促进海马CA1谷氨酸释放,而持续增加Aβ浓度后这种刺激作用会减弱[27-28]。这提示,在认知功能障碍出现之前,低浓度的Aβ可能已经诱发谷氨酸释放增多。由于研究手段的限制,无法直接向临床患者大脑输送钾离子来刺激谷氨酸释放,但是临床研究发现,脑脊液Aβ42阳性的健康老年人,胶质细胞功能标记物mIn的表达量明显高于脑脊液Aβ42阴性健康老年人[29],这说明Aβ42的表达可能是导致胶质细胞功能增强的原因之一。对APP转基因鼠的研究也发现,8月龄小鼠未检测到胶质化和老年斑形成,TBOA抑制谷氨酸重吸收实验发现皮层谷氨酸重吸收活力增强,细胞外谷氨酸浓度下降。18月龄APP转基因小鼠大脑出现老年斑形成和胶质化现象,皮层中胶质细胞重吸收功能相关的高亲和力谷氨酸转运体1(glutamate transporter 1,GLT1)显著下降,皮层细胞外谷氨酸浓度呈现升高趋势[30]。这些研究提示,在发生明显的认知功能障碍之前,有可能存在一个漫长的认知损伤阶段,并且在这一阶段,谷氨酸的诱发释放增多,胶质细胞功能增强。
进入轻度认知功能障碍(mild cognitive impairment,MCI)阶段后,AD病理改变加重,临床研究发现,MCI患者脑内谷氨酸水平与对照组无异或仅有轻微下降,而AD患者脑内谷氨酸水平显著下降[31-32]。对正常年老者,MCI及AD患者进行大脑基因表达谱研究显示,MCI患者的内嗅皮层、海马、额上回、中央后回四个脑区中的突触囊泡和释放相关蛋白的表达是升高的。临床前AD患者仅在老年斑组织处出现VGLUT1的表达明显下降,在无老年斑处没有出现VGLUT1的表达下降,12月龄APP/PS1小鼠的无老年斑脑区甚至出现VGLUT1的表达升高,而AD患者脑组织中不论是老年斑组织还是老年斑周围及无老年斑组织处均呈现出谷氨酸转运体VGLUT1的表达下降[33]。对CD1小鼠的研究也发现,12月小鼠出现认知功能下降(水迷宫),背侧海马中SNAP25及Munc18-1的表达量明显上升[34-35]。这些证据提示一种MCI状态下谷氨酸释放增多的现象,而当疾病进展至AD阶段后,这些递质释放相关蛋白表达会进入下降阶段[36]。复杂的是,不同脑区在疾病进程中的损伤并非同步,例如后扣带回作为一个AD疾病进程中较早损伤的脑区,在MCI阶段突触前蛋白synapsin-1及synaptophysin的表达就已经出现显著的下降[37]。除此之外,研究发现在AD患者脑内,促进谷氨酸合成的两种酶类,谷氨酰胺酶和谷氨酰胺合成酶的表达均显著下降[38-39],说明AD患者谷氨酸合成能力减弱,而AD动物模型研究发现,在AD早期,谷氨酰胺合成酶的表达并没有明显受累[40]。
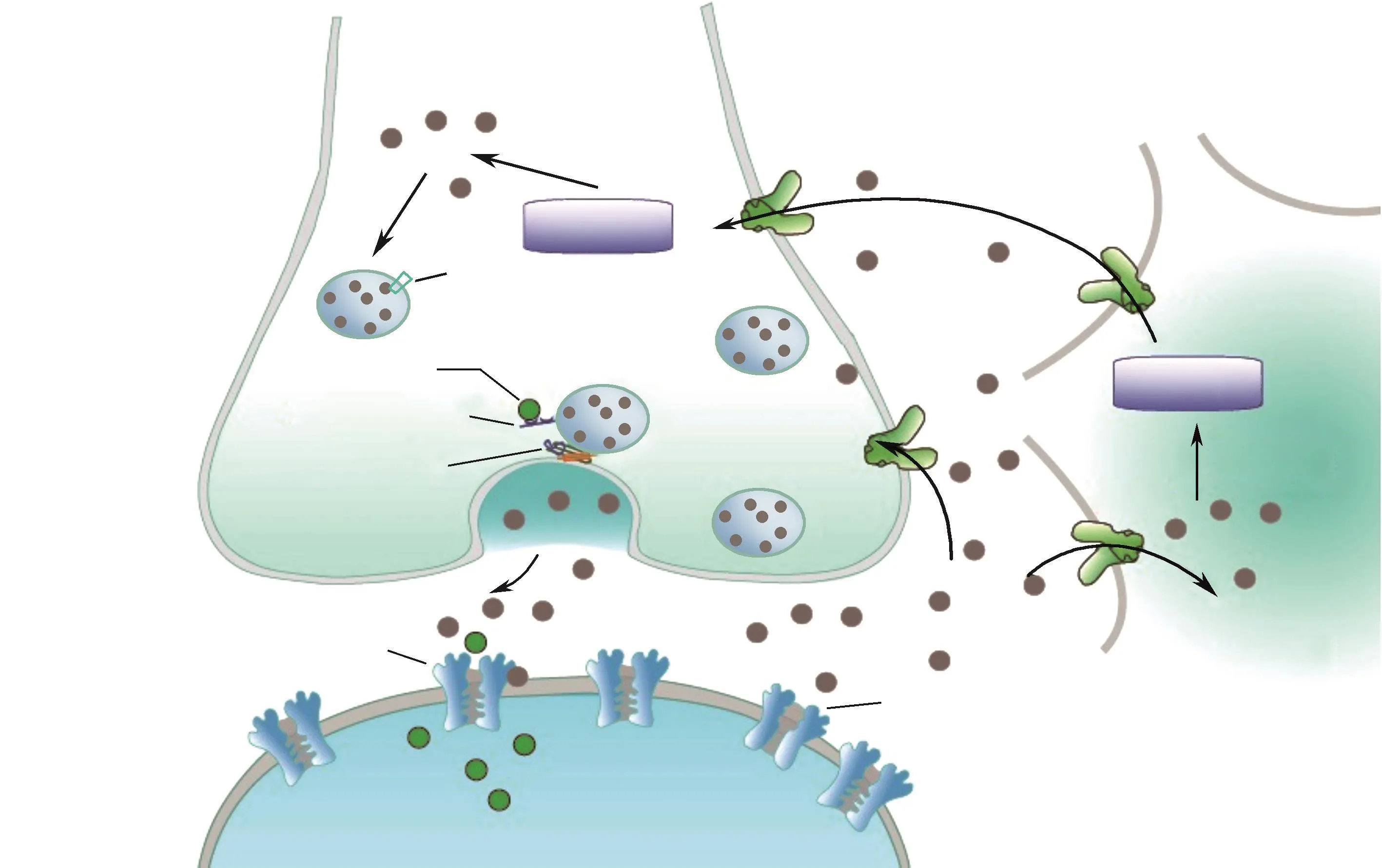
Fig.1 Process of glutamate synthesis,release and reuptake
由此可见,虽然在疾病向MCI和AD进展的过程中,大脑中谷氨酸的总量呈现出逐渐降低的变化趋势,但是在MCI阶段,谷氨酸的释放可能是增加的,因为众多神经递质释放相关蛋白的表达量是升高的,而此时胶质细胞的功能增强,导致细胞外谷氨酸浓度不变或略有减少。当疾病进展至AD阶段以后,递质释放相关蛋白减少,然而胞外Aβ可以增加囊泡释放的几率,谷氨酸引起的突触活动亦可以通过增加α-分泌酶向β-分泌酶的转变从而增加胞外Aβ浓度。可溶性Aβ寡聚体在突触损伤和神经退行性变的发生机制中起重要的作用[36],高水平的Aβ寡聚体可以使谷氨酸释放概率增加并阻滞神经元在突触间隙对谷氨酸的重吸收,其可能的机制是还原型辅酶Ⅱ(nicotinamide adenine dinucleotide phosphate,NADPH)氧化酶产生的活性氧和脂质过氧化产物造成的氧化性损伤抑制了谷氨酸转运蛋白,使谷氨酸水平升高,增多的谷氨酸一部分可以激活N-甲基-D-天冬氨酸(N-methyl-D-aspartic,NMDA)受体,随后使受体的敏感性降低,一部分可以发生外溢并激活突触外的富含NR2B亚基的NMDA受体。NMDA受体被过度激活后可诱导大量Ca2+内流,使线粒体Ca2+超载,细胞膜去极化而造成神经元功能障碍和细胞死亡[41]。
根据以上研究,在患者出现认知功能损伤之前,神经系统已出现功能异常,谷氨酸释放增加,胶质细胞功能增强,谷氨酸回收代偿性增加以减弱由谷氨酸释放增加带来的损伤。随着疾病进展,细胞外谷氨酸浓度下降,患者出现临床症状。至AD阶段,虽然脑内谷氨酸含量下降,却在Aβ的作用下过度释放,间隙谷氨酸浓度升高,产生谷氨酸兴奋性毒性。但是在AD最初阶段究竟是神经元损伤导致神经胶质细胞代偿活动增加,还是神经胶质细胞活动增强诱发神经元代偿性释放谷氨酸增多其实还并不清楚,有待更多的研究证实。
2 谷氨酸的突触后功能与AD
2.1 谷氨酸的生理功能
作为中枢神经系统中主要的兴奋性神经递质,谷氨酸需作用于突触后谷氨酸受体发挥功能。谷氨酸受体包括离子型和代谢型受体,离子型谷氨酸受体主要包括三类:NMDA受体,AMPA(α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid)受体和KA(kainic acid)受体;代谢型谷氨酸受体(metabotropic glutamte receptor,mGluRs)系属G蛋白耦联受体。三种离子型受体结构组成的不同之处在于:NMDA受体主要含有七种亚基,包括一个NR1亚基,四个NR2亚基(NR2A,NR2B,NR2C和NR2D)以及两个NR3亚基(NR3A,NR3B)[42-43],其中NR1为功能性亚基。NMDA受体在海马,丘脑和皮质中含量较多,其中在海马CA1区含量最多,而在基底节,小脑和脊髓中表达相对较少[44];AMPA受体包含GluR1-4亚基;KA受体包含GluR5-7及KR1-2亚基。谷氨酸受体在突触前,突触后神经元和胶质细胞中表达,谷氨酸在学习与记忆中的重要作用也通过其受体而发挥。
学习与记忆的神经基础是突触可塑性,而NMDA受体是经典的与学习与记忆相关的受体。NMDA受体的主要特点是其电压与配体双重门控性,在突触后膜去极化后,Mg2+与通道解离的同时谷氨酸与受体结合导致通道开放,一部分Ca2+通过NMDA受体耦联的Ca2+通道内流,一部分Ca2+通过电压门控的Ca2+通道内流,还有一部分Ca2+通过胞内释放从内质网进入胞浆[45],内流的Ca2+作为第二信使激活突触后神经元内各类激酶(CaMKⅡ、蛋白激酶C、蛋白激酶A、CAMKⅣ等),诱发并维持长时程增强(long term potentiation,LTP)[46]。
LTP的形成不仅依赖于NMDA受体,AMPA受体和KA受体也发挥了重要作用[47]。谷氨酸同时作用于NMDA,AMPA及KA受体,但由于NMDA受体受Mg2+阻滞作用而处于非活化状态,只有当AMPA受体被激活后,Na+内流导致突触后膜发生去极化,NMDA受体才能被激活并介导Ca2+内流[48]。除离子型谷氨酸受体外,mGluRs在学习与记忆的形成过程中也发挥重要作用,mGluRs属于G蛋白耦联受体超家族[49]。mGluRs在长时程抑制(long term depression,LTD)中的作用尤为重要,突触传递中的LTD可由低频刺激诱导产生,且LTD的维持依赖于NMDA受体和mGluRs的作用。但是也有研究表明,阻断NMDA受体并不会影响LTD的产生,而同时应用mGluRs的拮抗剂则阻断LTD的产生,说明mGluRs功能对LTD的诱导至关重要[50]。谷氨酸及其受体基本功能以及其在海马及皮层局部环路中的作用已经研究的相当深入,目前的研究发现,许多大脑远程投射纤维也可以共释放或调节谷氨酸功能[51-52],而这一部分谷氨酸功能调控还需更多探讨。
2.2 突触后谷氨酸功能异常与AD
作为AD主要的病理改变,Aβ的沉积对谷氨酸介导的突触可塑性有显著影响。在体及离体研究表明,可溶性Aβ寡聚体可以抑制LTP的形成,而易化LTD的产生[53-54]。作为参与突触可塑性形成的重要环节,NMDAR、AMPAR以及mGluRs功能均有可能受到Aβ的调节。APP转基因动物研究表明,LTP的损伤在Aβ斑块形成之前便已出现[55-56],向大鼠脑内注射可溶性Aβ寡聚体也可以特异性的损伤海马LTP及大鼠认知功能[57-58],这一点与突触前损伤是相对应的。科学家们曾经指出,在神经元出现丢失之前,突触功能的损伤是导致认知功能损害的主要原因[59]。对AD患者大脑的研究发现,AMPA受体结合位点显著减少[60-61],说明AMPA受体的表达量可能出现减少。在体及离体研究发现,Aβ的暴露会导致Caspase活性增强以及AMPA受体亚基的裂解。然而Caspase活性的增强并没有引起NMDA受体亚基的裂解[62]。因此在AD早期,Aβ的升高引发的AMPA受体裂解作用可能参与了突触传递障碍的过程中。而谷氨酸受体间的这种差异性调控可以成为日后药物研发过程中特别关注的问题。
除与AMPA受体相互作用外,Aβ与NMDA受体之间的关系似乎更为复杂,有研究指出,在AD起始阶段,Aβ的升高可能减少膜表面NMDA受体的表达量[63]。NMDA受体与突触后密度蛋白PSD95结合形成的复合体就是Aβ介导其突触后毒性作用的方式之一[64],Aβ可以通过STEP61(striatal-enriched protein tyrosine phosphatase 61)促进GLUN2B亚基Tyr1472位点的去磷酸化[65],介导NMDAR内化。可溶性Aβ寡聚体还可以通过作用于EphB2受体蛋白(ephrin type-B receptor 2),破坏NMDA受体的完整性,从而影响学习与记忆功能[66]。
然而复杂的还不仅于此,离体向海马神经元直接给予Aβ后,AMPA电流受到抑制,而NMDA电流却显著增强[67],而NMDA受体的激活还可以促进α分泌酶向β-分泌酶的转变,从而增加Aβ的生成[68],由此形成一种恶性循环。除此以外,可溶性Aβ寡聚体导致的含GluN2B亚基的NMDA受体介导的钙离子内流增加,还会激活下游信号通路如Rap-p38-MAPK、蛋白磷酸酶PP1、calcineurin。PP1可通过脱磷酸化激活GSK3β,同时影响Caspase-Akt通路,促进AMPA受体内化[69-71]。Calcineurin的激活则抑制AMPA受体ser-845位点磷酸化,促进AMPAR内化作用[72]。除促进AMPAR内化外,Aβ还可以通过引发关键激酶CAMKII的错误分布抑制AMPA受体的磷酸化和膜锚定过程[53]。mGLURs可与朊蛋白(prion protein)功能介导Aβ细胞内毒性作用[73-74]。在mGLURs介导的LTD过程中,AMPA受体的内化作用也受到STEP61的调节[75]。如此一来,Aβ通过促进AMPA受体内化和抑制其膜转移过程,减少膜AMPAR,抑制LTP及易化LTD的形成(Fig.2)。虽然Aβ与谷氨酸受体之间的相互作用关系已经研究的比较明确,但是值得注意的是,这些研究大多使用Aβ相关的转基因动物。然而对于临床前患者或者血管性痴呆疾患,Aβ沉积并不是其疾病发生的始动因素,因此对于AD发病早期谷氨酸功能的变化研究,还需要进一步细化。
Tau蛋白过度磷酸化引发神经纤维缠结是AD发病的另一重要假说[76]。Tau蛋白是一种微管相关蛋白,其功能主要为稳定微管及调节轴突运输。有研究表明,tau蛋白过度磷酸化对谷氨酸功能的损害作用主要体现在其可以介导Aβ的毒性作用[77-78],而tau蛋白敲除则可以减轻Aβ毒性作用[79-80]。此外,近年来AD炎症反应假说的研究受到研究者们的广泛重视[81-83]。中枢神经系统中炎症反应的主要参与者是小胶质细胞。目前,小胶质细胞与Aβ之间的调控关系比较多见[84-85]。但事实上,NMDA受体也表达与小胶质细胞上,但是其功能并不清楚[86]。在未来的研究中,谷氨酸功能与小胶质细胞之间的直接调控关系也是可以关注的方向之一。
3 谷氨酸通路中可能的AD治疗靶点
大脑中含有丰富的谷氨酸,但只有小部分的谷氨酸存在于细胞外。细胞内谷氨酸不具有兴奋性毒性,但是细胞外的谷氨酸被认为可以引发兴奋性毒性作用。根据大脑的能量需求和供应情况,谷氨酸不断从细胞中释放,又不断从细胞外空间中清除,使细胞外谷氨酸严格控制在低浓度。近年来,研究者们对于抑制脑内谷氨酸兴奋性毒性作用的潜在新型治疗方法产生较大的兴趣,各种治疗方案的核心在于将细胞外谷氨酸维持在合理的浓度范围内(Fig.3)。
由于细胞外谷氨酸浓度在AD发展不同阶段的波动变化,不同脑区在AD进程中退化程度也有所不同,针对谷氨酸通路的药物研发和使用会因其不同的波动状态而显得非常困难。仅予促进或抑制谷氨酸释放的药物也许并不能有效的缓解症状反而有可能对疾病的发展不利。柳叶刀杂志委员会也曾建议研究者“攻克AD重在预防”,如果能将AD治疗时间窗提前至认知功能异常之前,将事半功倍。但是在认知障碍出现之前谷氨酸诱发释放增多的机制还有待进一步探讨。在病人出现认知障碍之前,可通过调节谷氨酸释放而避免进入认知障碍阶段,可能的方案包括抑制递质释放相关蛋白如SNARE复合物,synaptotagmin的功能[87-88]。我们还可以屏蔽Aβ对于谷氨酸释放的增强作用,例如屏蔽Aβ与α7烟碱受体的作用位点[89-90]。研究发现,Aβ可以通过α7烟碱受体调节谷氨酸释放[91-92],Aβ还可以通过α7烟碱受体刺激星形胶质细胞释放过量的谷氨酸。这样一来,通过调节Aβ与α7烟碱受体之间的相互作用也许可以达到间接调节突触间隙谷氨酸浓度的作用。除此以外,在MCI阶段,通过抑制星形胶质细胞功能,减少对谷氨酸的重吸收作用,增加细胞外谷氨酸浓度,也是一个可以尝试的方案。也有研究发现,通过调节五羟色胺受体如5-HT6受体可以调控多种神经递质功能,如乙酰胆碱、谷氨酸、多巴胺等[93]。
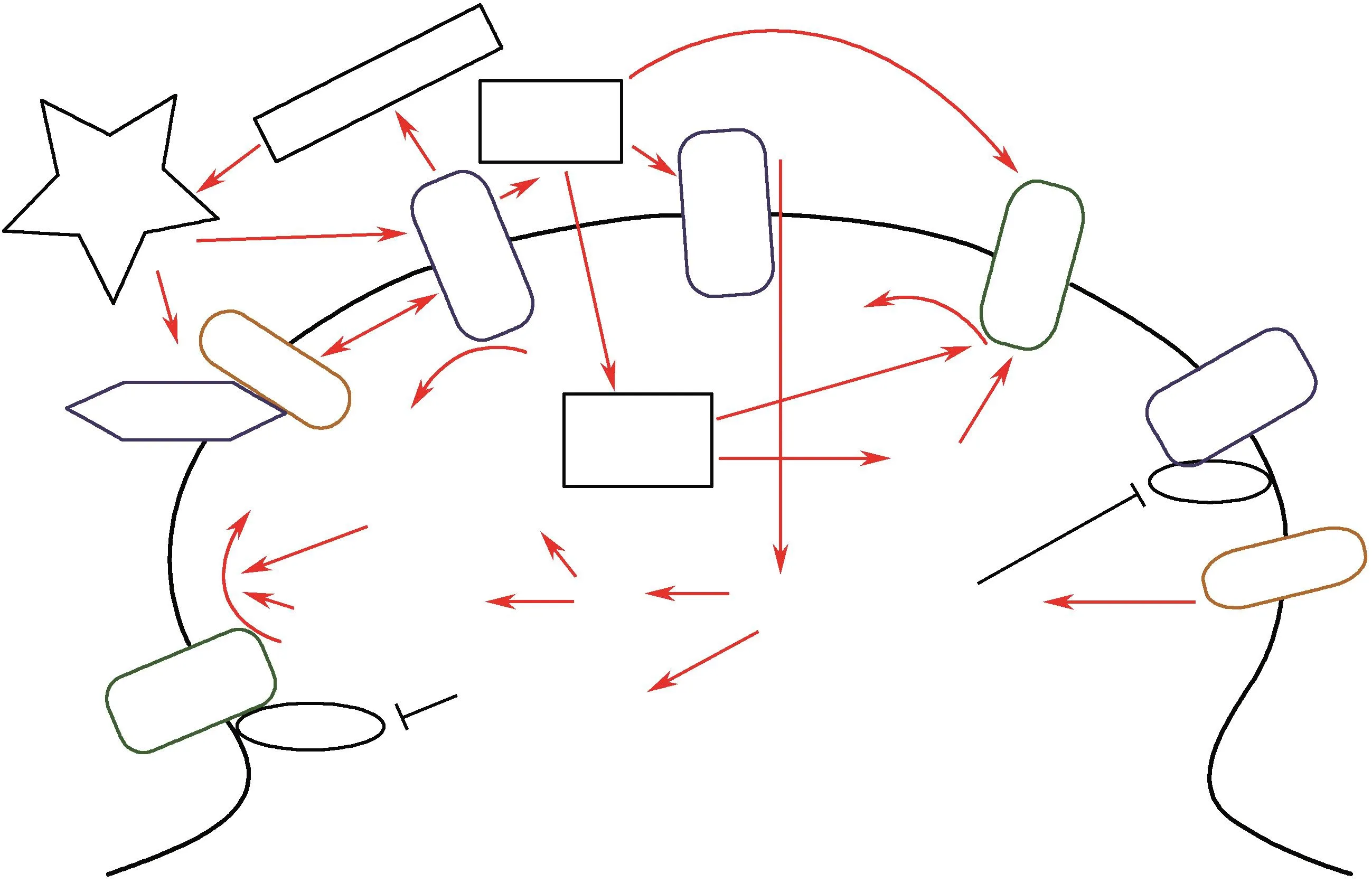
Fig.2 Interaction between Aβ and glutamate receptors (NMDARs, AMPARs and mGluRs)
当疾病进展至AD以后,谷氨酸循环障碍更为显著。针对谷氨酸功能的AD治疗药物目前只有非竞争性NMDA受体拮抗剂美金刚。NMDA受体具有较强的电压依赖性,这使得美金刚在阻止病理性Ca2+内流和突触后神经元的氧化应激效应的同时还能够保持较强的生理信号传递的能力。目前,美金刚已被证明还可以保护神经元免受线粒体毒性和缺氧的损害作用[94-95]。此外,研究发现美金刚在AD的临床前阶段也可以发挥有益作用。长期给予美金刚可以使Aβ斑块的沉积显著降低,突触密度升高并使退行性轴突减少[96]。这说明美金刚不但可以改善AD临床前阶段的记忆衰退,而且可以缓解晚期老年痴呆中的认知缺陷[97]。近期研究发现,NMDA受体拮抗剂Radiprodil也可以逆转Aβ对LTP的损伤[98]。STEP61的小分子抑制剂TC-2153可抑制AMPA受体内化有效逆转AD小鼠的认知功能损伤[99],恢复EphB2表达也可以有效恢复AD小鼠的认知功能[100]。谷氨酸调节因子riluzole可以逆转衰老和AD基因表达谱[101],mGluR Ⅱ拮抗剂可以缓解AD动物记忆损伤及焦虑情绪[102]。近年来非编码RNA研究领域发展迅速,长链非编码RNA(lncRNA),小RNA(miRNA)[103],环状RNA(circRNAs),都具有多靶点调控的功能特点[104],也许未来以非编码RNA为基础的药物会进入我们的视线。
谷氨酸与受体结合后会立即被清除或重吸收以维持突触间隙中谷氨酸的低浓度,有研究显示在AD中EAAT的活动受到抑制,导致谷氨酸清除减少,谷氨酸转运体受到Aβ的损伤,这是由于Aβ可以产生4-羟基-2-壬烯醛抑制EAAT的活动,导致细胞外谷氨酸清除障碍[105],使其在突触间隙堆积引起兴奋性毒性,因此在AD阶段调节EAAT的活性,同时抑制胶质细胞释放谷氨酸也可能成为有效的治疗手段[106-107]。
除以上的方案以外,考虑到Aβ沉积和tau蛋白磷酸化的影响,可以采用特异性抗体进行治疗,但是曾被寄予厚望的Solanezumab临床试验效果并不理想[108]。2002年至2012年间,有244种化合物先后进入AD治疗临床试验,但仅美金刚获得FDA批准[109]。目前,Aβ抗体、β和γ分泌酶抑制剂的临床试验纷纷失败,纠其原因可能是由于干预的时间点过晚,Aβ斑块已经形成,若能在斑块形成之前给予干预,避免斑块形成而诱发神经损伤,或许会得到更为理想的治疗效果。研究发现,在AD早期,Aβ抗体可有效缓解AMPA受体功能损伤及细胞骨架完整性受损[110]。最近,抗β-淀粉样原纤维抗体BAN2401在一项2期临床试验中,取得了积极的顶线成果,在预先设定的临床终点上(18个月),显著延缓了AD的进展评分,也减少了大脑中淀粉样蛋白的积聚[111-112],为以Aβ为靶点的AD药物治疗带来了新的希望。另外,光遗传学的发展也为谷氨酸功能的精准调控以及神经环路调控提供了新的契机[51,113]。
4 展望与小结
虽然大量的研究为我们提供了众多的药物靶点,但是在针对谷氨酸通路的药物研发中有一些重要的问题是不容忽视的。例如,递质释放相关蛋白不仅仅涉及谷氨酸释放,还参与GABA,多巴胺,肾上腺素等神经递质的释放[114-117]。如果简单抑制递质释放相关蛋白的表达,对于情绪的影响是无法预测的,对于神经递质调节类药物的研发,以及靶点的选择有赖于科学家们对神经递质间相互调控关系更为深入的理解。另外,从MCI进展至AD阶段,细胞外谷氨酸浓度会经由先降低后升高的过程,这使得调节谷氨酸重吸收功能的时间点比较难以把控,需要依赖于精准的诊断技术。最后,突触前神经递质的合成和释放是一个非常复杂的过程,单一因素的调控也许并不能帮助我们达到理想的治疗效果,因此寻求可以多靶点调控神经递质合成、释放或重吸收功能模块的潜在药物也许有重要的意义。
谷氨酸能神经传递的调控是一个复杂而微妙的平衡过程,谷氨酸功能亢进或减弱都会引起神经元功能障碍甚至痴呆发生,因此了解谷氨酸通路在痴呆进程中的病理变化过程对于预防和治疗阿尔茨海默病大有裨益。
