ATG5介导肝癌细胞对抗营养危机的作用研究*
2018-06-14马成马孙强李济宇
马成,马孙强,李济宇
(1.安徽医科大学上海临床学院,上海 200072;2.上海市第十人民医院,上海 200072)
自噬是细胞内最重要的house-keeping的功能之一。自噬通过依赖溶酶体降解或消化细胞内受损的器官和蛋白或入侵微生物来获取再生的能量:先围圈这些物质形成双膜的隔间被称为“自噬体”之后被溶酶体消化。当细胞面对外源压力比如营养缺乏时,启动自噬补充细胞所需的营养是细胞生存的重要手段[1-2]。
自噬和人类疾病的关系是近年来的研究热点。研究已经明确自噬是人体引发免疫功能和清除病原体所必需的[3-6]。自噬是细胞生存的重要途径,但其与细胞死亡的关系不清楚[7]。尤其是肿瘤细胞的自噬以及其和对抗营养缺乏的分子途径了解很少[8-10]。
研究表明,ATG5是细胞自噬途径中非常关键的蛋白。自噬体形成中需要二组接合(conjugation)系统(ATG12和ATG8)的协同作用,这二组接合系统都需要ATG5的参与[11-12]。缺乏ATG5导致小鼠B细胞发育不全[13],小鼠出生后立刻死亡[14-15]。
p53是细胞中最负盛名的肿瘤抑制因子,在60%以上人类肿瘤中发现该基因存在突变[16]。在细胞内,p53会诱导p21(cip1/waf1)这个下游产物的表达来抑制细胞增殖[17-18]。p21(cip1/waf1)是细胞中重要的细胞周期的负调控蛋白质(CDKI)。通过与细胞周期中的cyclins/CDKs形成复合物从而抑制Rb的磷酸化从而使细胞停留在G1或者G2期[19-20]。现在有很多研究证明,p21(cip1/waf1)在很多生物环境中帮助肿瘤的生存。
本研究将小鼠正常肝细胞AML-12、肝癌细胞Hepa 1-6在缺血条件下进行培养,探讨自噬基因ATG5在肿瘤细胞中对抗营养危机的作用。
1 材料与方法
1.1 材料
鼠细胞系AML-12和Hepa 1-6(中国科学院典型培养物保藏委员会细胞库),胎牛血清、DMEM/F12培养液、1000×青霉素/链霉素溶液及0.25%含EDTA胰酶(美国Gibico公司),Lipofectamine®3000试剂、二氧化碳CO2无菌培养箱和实时荧光定量聚合酶链反应(quantitative real-time polymerase chain reaction,qRT-PCR)仪(美国Thermo公司);ATG5、p21小干扰 RNA(si-ATG5,si-p21)、对照小干扰 RNA(si-con)及ATG5过表达质粒(上海捷瑞生物工程有限公司构建),RNAiso plus、逆转录试剂盒及SYBR®Green染料(大连宝生物工程有限公司),定量引物(上海捷瑞生物工程有限公司),碘化丙啶(PI)、RNase A、Triton X-100、RIPA裂解液及CCK-8试剂盒(上海碧云天生物技术有限公司),兔抗人ATG5单克隆抗体、兔抗人p21单克隆抗体、鼠抗人GAPDH单克隆抗体及抗鼠兔二抗(美国Abcam公司),PVDF膜(德国merckmillipore公司),酶标仪(美国Biotek公司),Odyssey双色红外荧光成像系统(美国LI-COR公司),流式细胞仪(美国BD biosciences公司)。
1.2 方法
1.2.1 细胞株和细胞培养 小鼠的AML-12和Hepa 1-6细胞于37℃、5% CO2培养箱中培养,覆盖率达80%~90%进行细胞传代;实验过程所需材料(血清和培养基除外)均需放置在超净台上并用紫外灯照射30 min灭菌,先用酒精棉球擦拭培养瓶瓶盖,打开后瓶口在酒精灯上过火焰,再用吸管轻轻吸出旧的培养液至废液缸,加入2 ml无菌PBS洗1次;加入1 ml的2.5%胰蛋白酶-EDTA消化液于37℃消化细胞数分钟。倒置显微镜观察到细胞突起消失变圆后,加入等体积的培养基(10% FBS+100 u/ml青霉素+100 u/ml链霉素+90% DMEM/F12培养基)终止消化。用移液枪吹打细胞,使其悬浮,将细胞吸到15 ml的离心管中,用培养基清洗培养瓶合并入离心管,1 000 r/min 5 min。弃上清液至废液缸,加入1 ml培养基,将细胞混匀成细胞悬液,以1∶2或1∶3进行传代,放入培养箱中培养,当细胞处于对数生生长期时,进行缺血处理或者其他后续实验。对处于对数生长期的细胞进行缺血培养实验,将培养基换成90% DMEM/F12培养基+100 u/ml青霉素+100 u/ml链霉素的无血清培养基放入培养箱中培养,24 h后进行后续实验作为研究组。以90% DMEM/F12培养基+100 u/ml青霉素+100 u/ml链霉素的含血清培养基放入培养箱中培养,作为对照组。
1.2.2 转染质粒及si-RNAs 细胞转染应用Lipofectamine ®3 000试剂,按照用户手册进行转染。转染质粒时6孔板每孔5 μl Lipofectamine®3 000+5 μl P3 000转染试剂+2 500 ng目的质粒,转染si-RNA时6孔板每孔5 μl Lipofectamine®3 000+75 pmol目的si-RNAs。
1.2.3 细胞的RNA提取和qRT-PCR分析 待细胞转染24 h后,收集各组细胞,用TaKaRa公司的RNAiso plus试剂按照操作手册提取细胞总RNA;取1 μg RNA逆转录得到cDNA,用SYBR Green染料检测ATG5或p21 mRNA的表达水平。ATG5 mRNA正向引物:5'-GAAAGAGTGTGTCCTCCTCG-3',反向引物:5'-TTGCCTCCACTGAACTTGAC-3';p21正向引物:5'-CTTGCACTCTGGTGTCTGAG-3',反向引物:5'-CTGCGCTTGGAGTGATAGAA-3';内参引物 GAPDH正向引物:5'-GCAAAGTGGAGATTGTTGCC-3',反向引 物:5'-TTGAATTTGCCGTGAGTGGA-3'。ATG5或p21 mRNA转录水平的相对表达量用2-ΔΔCt法分析。
1.2.4 CCK-8法检测转染后细胞增殖能力变化 胰酶消化对数生长的细胞,收集细胞并进行细胞计数,每孔种3×103个细胞于96孔细胞板内每组设6个复孔,在每组实验细胞周围的孔加10 μl PBS于7℃、5%CO2培养箱中培养。细胞缺血培养或者转染24 h后检测细胞增殖活性,检测前在每个待检测孔加入10 μl CCK-8,将细胞板放入培养箱中继续孵育2 h,在全自动酶标议上测定各孔吸光度值(450 nm为测定波长,630 nm为参比波长)。细胞生长抑制率=(1-实验组A值/对照组A值)×100%。
1.2.5 Western bolt检测蛋白表达 收取各组细胞,用冰浴的PBS洗涤2次;加入RIPA裂解液收取总蛋白,测定蛋白浓度;将30 μg蛋白经聚丙烯酰胺凝胶电泳后,转移至PVDF膜;5%脱脂奶粉室温封1 h,4℃下结合ATG5、p21或GAPDH(内参)一抗过夜;PBST洗3次,室温结合二抗1 h;PBST洗3次,Odyssey显影,凝胶成像仪对蛋白条带进行灰度分析,计算目的蛋白与内参蛋白条带的灰度值比值。
1.2.6 细胞周期实验 细胞转染后24 h后每样收集1×106个细胞,离心收集细胞,弃上清液,用预冷PBS洗细胞2次,加入预冷70%乙醇,于4℃固定过夜,或置入-20℃长期固定。离心收集细胞,以1ml的PBS洗细胞1次,加入500 μl PBS含50 μg/ml碘化丙啶(PI),100 μg/ml RNase A,0.2% Triton X-100,4℃避光孵育30 min。以标准程序用流式细胞仪检测,一般计数2万~3万个细胞,结果用细胞周期拟和软件ModFit分析。
1.3 统计学方法
数据分析采用SPSS 17.0统计软件,计量资料以均数±标准差(±s)表示,用单因素方差分析,若方差齐,则两两比较用LSD-t检验,P<0.05为差异有统计学意义。
2 结果
2.1 两种细胞缺血培养的增殖情况
缺血24 h后AML-12细胞和没有全营养的对照比较,细胞的死亡增加近10%;而Hepa1-6的24 h缺血导致的死亡增加约3%,经t检验,差异有统计学意义(P<0.05)。见表1。
表1 两种细胞缺血处理后与对照细胞的死亡率比较(n =6,%,±s)

表1 两种细胞缺血处理后与对照细胞的死亡率比较(n =6,%,±s)
组别 AML-12 Hepa1-6对照组 2.488±0.652 1.888±0.584研究组 12.163±1.056 4.758±0.425差值 9.675±0.404 2.870±0.159 t值 19.091 8.031 P值 0.000 0.000
2.2 两种细胞缺血对内源ATG5表达的影响
与对照组比较,缺血24 h后AML-12细胞内的自噬相关基因ATG5的表达升高约1倍;而Hepa1-6细胞缺血24 h导致ATG5基因表达升高近4倍,经t检验,差异有统计学意义(P<0.05)(见图1A和表2)。Western blot的结果显示,缺血24 h后AML-12细胞内的自噬相关蛋白ATG5的表达升高达1.6倍。而Hepa1-6细胞的缺血24 h导致ATG5蛋白表达升高达4.5倍(见图1B)。
2.3 转染si-RNAs、质粒后Hepa1-6细胞的增殖情况
转染si-RNAs后,与对照组比较两种细胞ATG5蛋白表达降低60%以上(见图2A)。转染si-RNAs后两种细胞缺血培养24 h,与对照组比较,AML-12细胞的死亡未增加(P>0.05);而Hepa1-6细胞死亡升高13%,经t检验,差异有统计学意义(P<0.05)(见图2B和表3)。转染外源ATG5基因表达的质粒后,与对照组比较,ATG5蛋白表达升高达4.3倍(见图2C)。转染质粒后Hepa1-6细胞缺血培养24h,细胞死亡百分率为(2.413±0.601)%,与对照组(5.542±0.838)%比较,降低3.1%,经t检验,差异有统计学意义(t=7.168,P=0.000)(见图2D)。
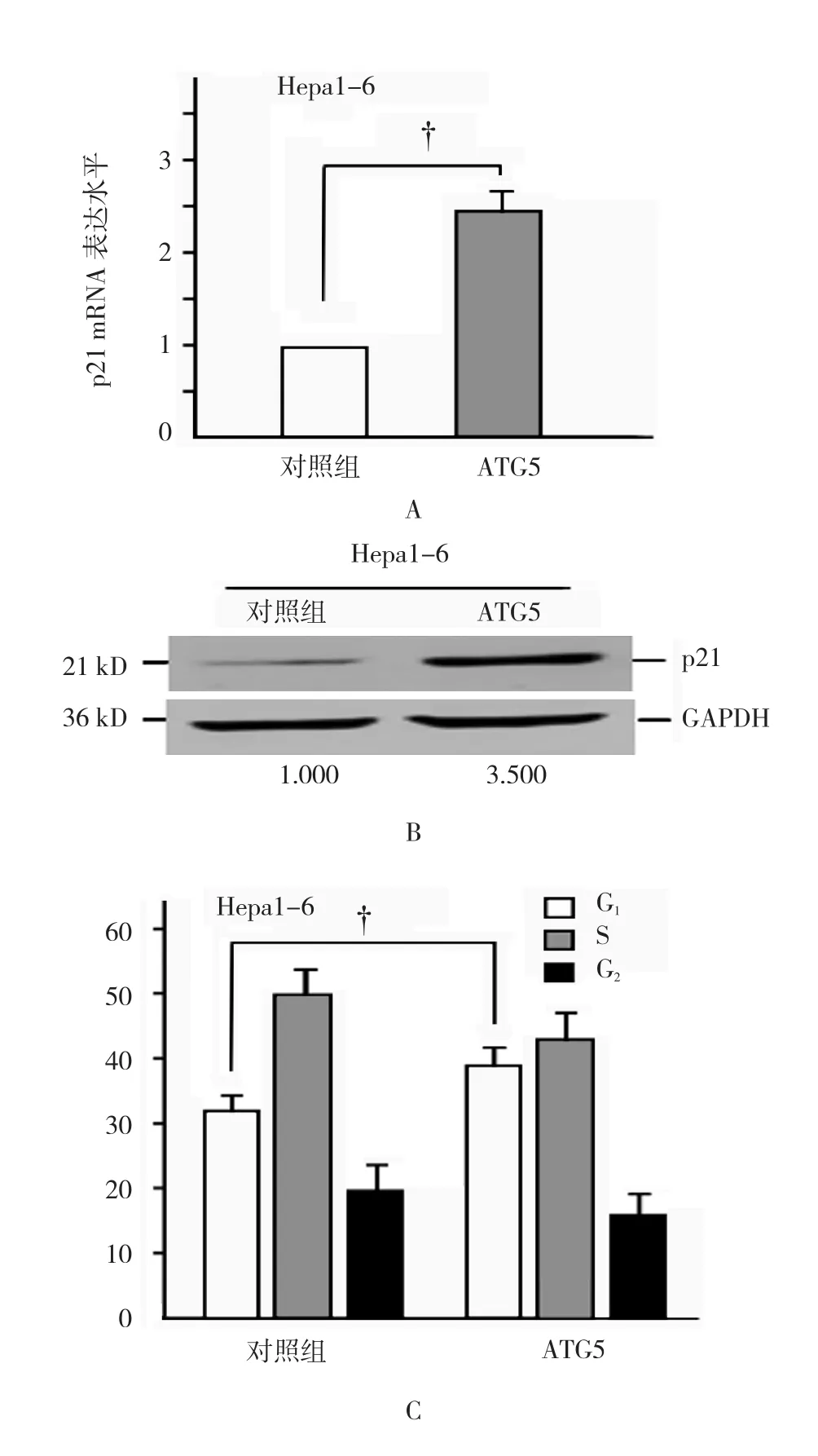
2.4 ATG5对细胞周期抑制蛋白p21(cip1/waf1)的影响
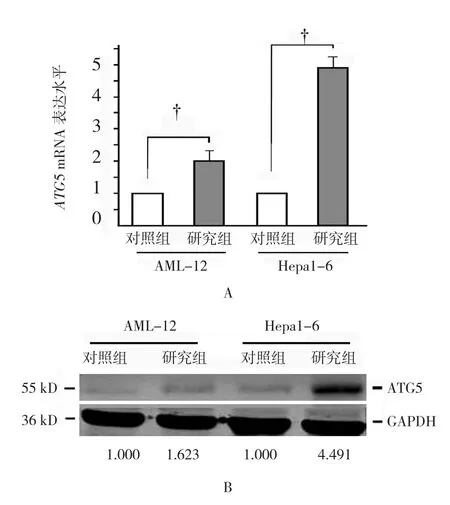
图1 两种细胞缺血处理后对内源ATG5表达的影响
表2 两种细胞缺血处理后ATG5的表达比较(n =6,%,±s)
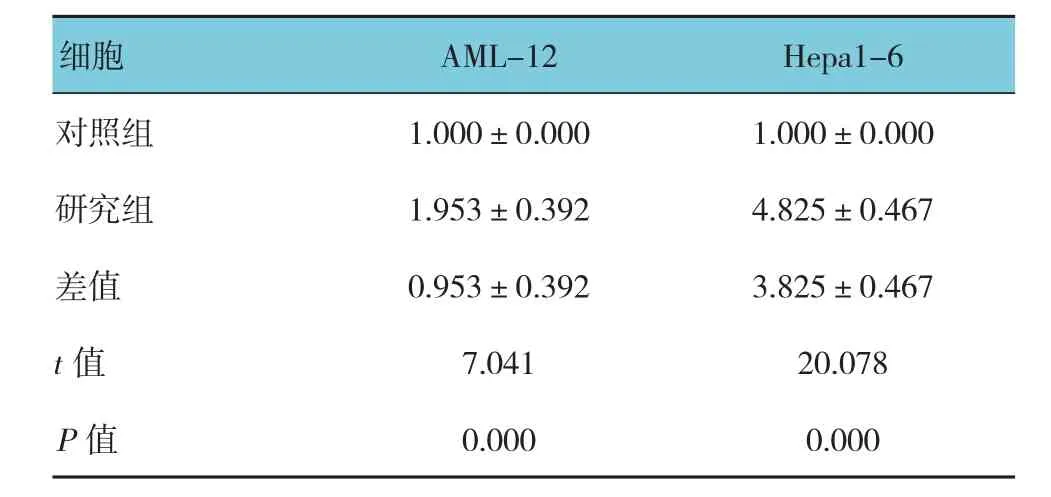
表2 两种细胞缺血处理后ATG5的表达比较(n =6,%,±s)
细胞 AML-12 Hepa1-6对照组 1.000±0.000 1.000±0.000研究组 1.953±0.392 4.825±0.467差值 0.953±0.392 3.825±0.467 t值 7.041 20.078 P值 0.000 0.000
转染外源ATG5表达的质粒后,p21(cip1/waf1)mRNA表 达(2.397±0.212)%,与对照组(1.000±0.000)%比较升高达2.4倍,经t检验,差异有统计学意义(t=16.173,P=0.000)(见图3A)。细胞内源p21(cip1/waf1)蛋白表达升高达3.5倍(见图3B)。流式细胞仪测定显示,Hepa1-6细胞的细胞周期G1期从空载体的(31.467±1.583)%升高到ATG5转染的(38.067±1.780)%,经t检验,差异有统计学意义(t=6.787,P=0.000)(见图3C)。si-RNAs沉默p21(cip1/waf1)后p21蛋白的表达降低(见图3D)。转染si-RNAs后细胞缺血培养24h,细胞死亡率(15.085±1.156)%,与对照组(4.792±0.609)%比较增加了10%,差异有统计学意义(t=19.297,P=0.000)(见图3E)。
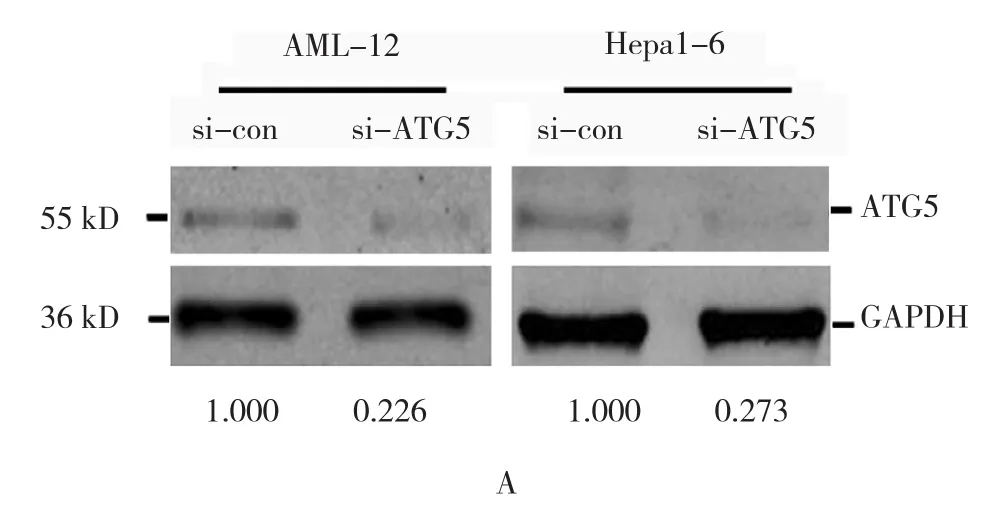

图2 转染si-RNAs、质粒后细胞增殖比较
表3 两种缺血处理细胞下调ATG5后与对照组细胞的死亡率比较 (n =6,%,±s)
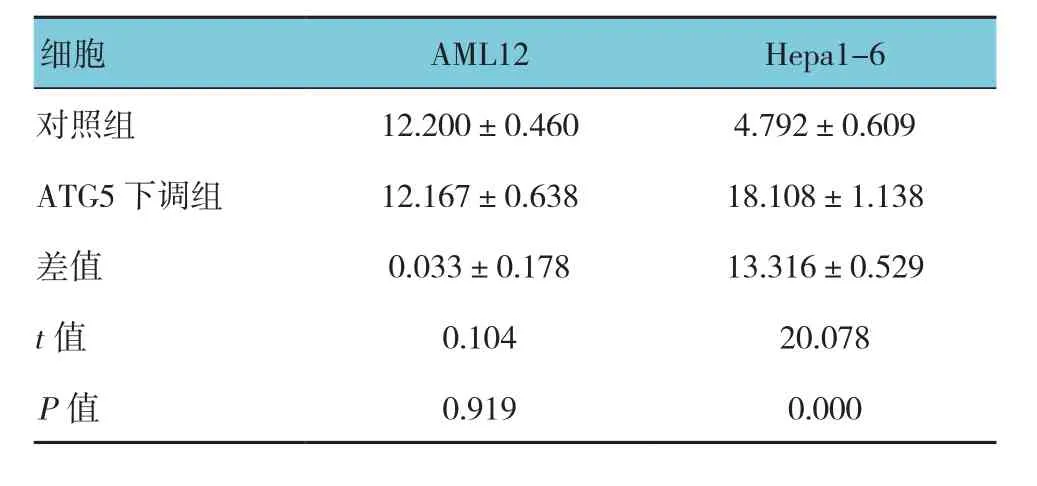
表3 两种缺血处理细胞下调ATG5后与对照组细胞的死亡率比较 (n =6,%,±s)
细胞 AML12 Hepa1-6对照组 12.200±0.460 4.792±0.609 ATG5下调组 12.167±0.638 18.108±1.138差值 0.033±0.178 13.316±0.529 t值 0.104 20.078 P值 0.919 0.000
3 讨论
肝脏作为体内最重要的代谢器官,本身可能会发生代谢失衡,特变是在慢性肝病中会伴有大量的肝脏细胞自噬和凋亡,自噬与凋亡的失衡也可导致肝脏疾病的发生,肝损伤的病理特征多为肝细胞的自噬与凋亡。研究发现,自噬失调与病毒性肝炎、非酒精性脂肪肝、酒精性肝病、纤维化、肝硬化和肝细胞癌有关[9-10],且肿瘤细胞依赖自噬在肝癌中存活[11]。因此研究自噬相关基因与肝脏疾病的关系对了解肝脏疾病显得尤为重要。
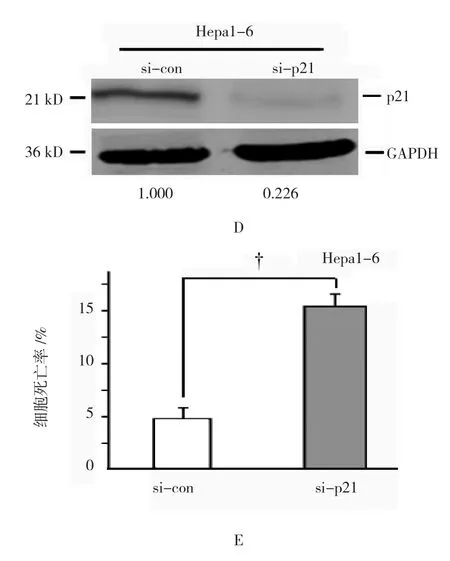
图3 ATG5对p21(cip1/waf1)的影响
本研究将小鼠正常肝细胞和癌细胞在不同条件下缺血培养,发现缺血导致一定程度上两种细胞内自噬蛋白ATG5升高,与前人的研究结果相符,即在营养缺乏的环境中细胞产生应激,启动自噬,自噬增强,以满足自身代谢需求[21]。且肝癌细胞Hepa1-6对缺血引起的细胞死亡有抗性。si-RNAs沉默ATG5基因后,可以提高肝癌细胞Hepa1-6对缺血的敏感性。在细胞外源表达ATG5蛋白情况下,会进一步降低细胞对缺血的不敏感性。且ATG5提高细胞内细胞周期的抑制蛋白p21(cip1/waf1)的表达,导致细胞滞留在G1期,可能以此逃避缺血带来的死亡危机。
自噬是细胞通过消耗自体而获得能量从来寻求生存的分子途径。肿瘤细胞的自噬和自噬的调节的研究还未完全清楚。许多研究也指出,营养剥夺可以诱导自噬并促进肝癌细胞增殖侵袭现象,但其根本的分子途径并不明确。本研究说明小鼠肝癌细胞通过升高自噬蛋白ATG5的表达来对抗生存环境中营养缺乏的危机。而且ATG5是通过提高细胞内细胞周期的抑制蛋白p21(cip1/waf1)的表达量,从而导致细胞滞留在G1期,可能以此来逃避缺血带来的细胞死亡危机。
自噬作为细胞内的一个重要代谢途径,其可以抑癌也可以促癌,但在治疗肿瘤过程中是促进自噬还是抑制自噬,还有待研究。但不可否认的是与正常组织比较,癌细胞更具有自噬依懒性,说明存在治疗窗口。本研究结果提示,自噬基因ATG5在肝癌细胞中发挥重要作用,ATG5帮助其对抗营养缺乏带来的生存危机,有望成为治疗进展期肝癌的潜在靶点。
[1]MIZUSHIMA N. The pleiotropic role of autophagy: from protein metabolism to bactericide[J]. Cell Death & Differentiation, 2005,12(Suppl 2): 1535.
[2]YORIMITSU T, KLIONSKY D J. Autophagy: molecular machinery for self-eating[J]. Cell Death & Differentiation, 2005, 2(12 Suppl):1542-1552.
[3]LEE H K, LUND J M, RAMANATHAN B, et al. Autophagydependent viral recognition by plasmacytoid dendritic cells[J].Science, 2007, 315(5817): 1398-1401.
[4]LEVINE B, DERETIC V. Unveiling the roles of autophagy in innate and adaptive immunity[J]. Nature Reviews Immunology,2007, 7(10): 767.
[5]TALLOCZY Z, JIANG W, LEVINE B, et al. Regulation of starvation- and virus-induced autophagy by the eIF2 alpha kinase signaling pathway[J]. Proc Natl Acad Sci USA, 2002, 99: 190-195.
[6]GUTIERREZ M G, COLOMBO M I, MASTER S S. Autophagy is a defense mechanism inhibiting BCG and mycobacterium tuberculosis survival in infected macrophages[J]. Cell, 2004,119(6): 753.
[7]LUM J J, BAUER D E, KONG M, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis[J]. Cell,2005, 120(2): 237.
[8]LIANG X H, JACKSON S, SEAMAN M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1[J]. Nature,1999, 402(6762): 672.
[9]VQU X, YU J, BHAGAT G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene[J]. Journal of Clinical Investigation, 2003, 112(12): 1809.
[10]MATHEW R, KONGARA S, BEAUDOIN B, et al. Autophagy suppresses tumor progression by limiting chromosomal instability[J]. Genes & Development, 2007, 21(11): 1367.
[11]MIZUSHIMA N, YAMAMOTO A B A, HATANO C M, et al.Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells[J]. Journal of Cell Biology, 2001,152(4):657-668.
[12]BURMAN C, KTISTAKIS N T. Autophagosome formation in mammalian cells[J]. Seminars in Immunopathology, 2010, 32(4):397-413.
[13]MILLER B C, ZHAO Z, STEPHENSON L M, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development[J]. Autophagy, 2008, 4(3): 309-314.
[14]KUMA A, HATANO M, MATSUI M, et al. The role of autophagy during the early neonatal starvation period.[J]. Nature, 2004,432(7020): 1032.
[15]HARA T, NAKAMURA K, MATSUI M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice.[J]. Nature, 2006, 441(7095): 885.
[16]AYROLDI E, PETRILLO M G, BASTIANELLI A, et al. L-GILZ binds p53 and MDM2 and suppresses tumor growth through p53 activation in human cancer cells[J]. Cell Death & Differentiation,2015, 22(1): 118-130.
[17]LUO Y, HURWITZ J, MASSAGUÉ J. Cell-cycle inhibition by independent CDK and PCNA binding domains in p21 Cip1[J].Nature, 1995, 375(6527): 159-161.
[18]CAO W, MA S L, TANG J, et al. A combined treatment TNF-alpha/doxorubicin alleviates the resistance of MCF-7/Adr cells to cytotoxic treatment[J]. Biochimica Et Biophysica Acta, 2006,1763(2): 182.
[19]LI R, SHOU W, HANNON G J, et al. Differential effects by the p21 CDK inhibitor on PCNA-dependent DNA replication and repair[J]. Nature, 1994, 371(6497): 534-537.
[20]LU Y, YAMAGISHI N, YAGI T, et al. Mutated p21WAF1/CIP1/SDI1 lacking CDK-inhibitory activity fails to prevent apoptosis in human colorectal carcinoma cells[J]. Oncogene, 1998, 16(6): 705-712.
[21]GONG L, DI C, XIA X, et al. AKT/mTOR signaling pathway is involved in salvianolic acid B-induced autophagy and apoptosis in hepatocellular carcinoma cells[J]. Int J Oncol, 2016, 49(6): 2538-2548.
