利用酵母双杂交系统从人胚胎肝脏cDNA文库中筛选与JNKK2相互作用的蛋白
2018-06-13孙杰常正尧张晓明王晶
孙杰,常正尧,张晓明,王晶
1.军事科学院 军事医学研究院 毒物药物研究所,北京 100850;2.解放军第302医院,北京 100039;3.空军军医大学,陕西 西安 710032
c-JunN 端激酶激酶 2(c-JunN-terminalki⁃nasekinase2,JNKK2)是 c-JunN端激酶(c-Jun N-terminalkinase,JNK)的上游激酶,也被称为丝裂原激活的蛋白激酶激酶7(mitogen-activated proteinkinasekinase7,MKK7),其调控着机体内众多生理进程,包括细胞增殖、分化、迁移和凋亡等[1-2]。既往研究及我们课题组前期的工作发现,JNKK2分子参与调控多种肿瘤的恶性增殖[3-6]。然而目前有关JNKK2在肝脏中的功能及调节机制研究还较少。酵母双杂交系统是一种可以快速、直接分析已知蛋白之间相互作用的常用方法,可用于分离与已知蛋白存在相互作用的配体,并明确其编码基因。该系统具有简便、高效、灵敏等特征,并能反映不同蛋白在细胞内的相互作用,常被用于基因的功能研究[7-8]。因此,为了探究JNKK2在肝脏中的调节机制,我们利用酵母双杂交系统,从人胎肝cDNA文库中筛选出能与JNKK2相互作用的蛋白,期望找到调控JNKK2-JNK信号通路的新分子,进而为更好地理解JNK信号通路在肝脏中的作用机制奠定基础。
1 材料与方法
1.1 材料
JNKK2KM质粒、GFP-JNKK2质粒由本室张纪岩教授提供[4];酵母菌株 AH109(MATa),诱饵载体质粒pGBKT7,文库载体质粒pGADT7,对照质粒 pGBKT7-53、pGBKT7-Lam、pGADT7-T、pCL1,预转化的人胚胎肝脏cDNA文库,营养缺陷型培养 基 ,X-α-gal,抗 GAL4DNA-BD 多 抗 购 自Clontech公司;YPDA培养板购自青岛海博生物科技有限公司;鲑鱼精DNA(CarrierDNA)、玻璃珠、3-AT、PEG3350、溶壁酶、苯甲脒、DMSO、PMSF、抗Flag标签抗体购自Sigma公司;无氨基酸酵母氮源(YNB)购自Difco公司;蛋白A/G琼脂糖珠购自SantaCruz公司;DNA聚合酶Pyrobest及其缓冲液、dNTPs购自TaKaRa公司;各种限制性内切酶及相应缓冲液购自NewEnglandBiolabs公司;其他化学试剂均为国产分析纯产品。
1.2 pGBKT7-JNKK2KM诱饵载体的构建与鉴定
根据JNKK2编码区序列设计两端分别带有EcoRⅠ、BamHⅠ酶切位点的特异性引物JNKK2-P5 (5′-CCGGAATTCATGGCGGCGTCCTCCCTGGA ACAG-3′)和 JNKK2-P3(5′-CGCGGATCCTTACTC AGTCTTCGCCATGACATC-3′)。以 JNKK2KM 质粒为模板,PCR扩增JNKK2KM编码区片段(PCR扩增条件:95℃预变性5min;94℃变性1min,56℃退火 40s,72℃延伸 90s,30个循环;72℃延伸10min)。PCR产物片段与pGBKT7空载体质粒经EcoRⅠ/BamHⅠ双酶切,用DNA连接酶于16℃连接,然后转化大肠杆菌DH5α,挑单克隆进行菌落PCR并提取质粒进行双酶切后,经12g/L琼脂糖电泳分析,对PCR及酶切鉴定均为阳性的克隆进行基因测序鉴定。
1.3 诱饵质粒的转化及表达鉴定
将pGBKT7-JNKK2KM转化AH109酵母菌(参照Clontech MatchmakerPretransformed Libraries UserManual),以AH109/pGBKT7和AH109空菌分别作为空载体对照和空白对照,涂布SD/-Trp或YPDA平板,置30℃培养箱中培养3~5d,从平板上分别挑取AH109/pGBKT7-JNKK2KM及AH109/pGBKT7转化的单克隆,接种至SD/-Trp液体培养基中,以未转化质粒的AH109菌落接种至YPDA培养液中,30℃、250r/min振荡培养20h,通过免疫印迹验证BD-JNKK2KM融合蛋白的表达[3-5]。
1.4 酵母双杂交系统从人胎肝cDNA文库中筛选与JNNK2相互作用的蛋白
挑取新鲜含诱饵基因的酵母菌落至50mL SD/-Trp培养基中,充分混匀,30℃、250~270r/min培养 16~20h至D600nm>0.8;1000r/min离心菌液 5 min,弃上清,用 4~5mLSD/-Trp培养基重悬,调整酵母细胞浓度,使终浓度大于1×108/mL。室温水浴解冻1支含预转化文库的Y187酵母菌液,将上述过夜培养的5mLAH109诱饵菌液与1mL预转化文库菌液在2L无菌烧瓶中混匀,加入2×YPDA/Kana培养基(含50g/mL卡那霉素),定容至 50mL;30℃、30~50r/min 培养 20~24h;离心收集交配液,沉淀用10mL0.5×YPDA/Kana培养基重悬。用无菌涂棒,按200μL/平板,均匀涂布交配液到 55块 SD/-Ade/-His/-Leu/-Trp(QDO)四缺陷选择培养板上(150mm),30℃恒温培养8~21d;同时按比例稀释交配液分别涂于SD/-Trp、SD/-Leu、SD/-Leu/Trp平板上(100mm),30℃恒温培养3~5d,计数菌落数,计算交配效率。
1.5 阳性克隆的分离与验证
将上述可见克隆分别用无菌牙签挑出,在SD/-Leu/-Trp平板上划线,分离单克隆;分别从每块SD/-Leu/-Trp板上挑取2个单克隆,涂在SD/-Ade/-His/-Leu/-Trp/X-α-Gal(加入合适浓度的3-AT)培养板上;30℃培养 48~72h;蓝色菌落为阳性结果,阴性结果呈粉红色;选取阳性克隆再重复分离培养2~3轮。取3轮均变蓝的阳性克隆,进行扩增、抽提酵母质粒(按照Tiangen酵母质粒提取试剂盒说明书),PCR鉴定。同时PCR阳性的克隆酵母质粒转化大肠杆菌DH5α宿主菌,涂在含氨苄青霉素(Amp)的LB培养板上,37℃倒置培养12h,挑取单克隆,用含氨苄青霉素的LB培养基培养,提取大肠杆菌质粒;用HindⅢ限制性酶酶切鉴定。
1.6 阳性克隆基因测序与分析
利用文库载体pGADT7对应的常规AD测序引物对获得的阳性克隆质粒进行测序,上游引物为 5′-CTATTCGATGATGAAGATACCCCACCAAAC CC-3′,下 游 引 物 为 5′-GTGAACTTGCGGGGTTTT TCAGTATCTACGAT-3′)。 测 得 的 DNA 序 列 用DNAMAN软件进行编辑,删除载体序列后剩余部分为插入文库基因;再利用NCBI网站BLASTN程序(http://www.ncbi.nlm.nih.gov/blastn)搜索同源序列并记录,优先选取同源性高且经过人工注释的序列;序列比对后,对相位正确的cDNA进一步分析编码蛋白的来源、功能等相关信息,去除公认的假阳性蛋白如线粒体蛋白、细胞骨架等。
1.7 免疫共沉淀验证阳性克隆与JNKK2蛋白的相互作用
挑取阳性克隆基因,构建到pcDNA3.1(-)真核表达载体上,用LipofectAMINE2000分别将1 μgORF60、GNB2L1、UBA3表达质粒及pcDNA3.1(-)空载体质粒转染293T细胞,同时共转1μg GFP-JNKK2质粒;用预冷的Co-IP裂解液裂解细胞,裂解液加入1μLM2抗Flag抗体及20μL蛋白A/G琼脂糖珠,在旋转混合仪上于4℃旋转过夜;用预冷的Co-IP裂解液洗蛋白A/G琼脂糖珠3~5次,弃上清;加入20 μL2×SDS上样缓冲液,进行蛋白电泳和免疫印迹检测。
2 结果
2.1 pGBKT7-JNKK2KM诱饵载体构建
构建好的载体经菌落PCR(图1A)及EcoRⅠ/BamHⅠ酶切(图1B)均能得到1200bp的JNKK2 DNA插入片段,测序后进行序列比对,发现与JNKK2KM基因序列相同,表明诱饵质粒pGBKT7-JNKK2KM构建成功。
2.2 诱饵AH109酵母菌重组子的鉴定
挑出诱饵酵母克隆,培养扩增后提取酵母质粒,PCR鉴定重组质粒,3个克隆在1200bp处均有JNKK2KM目的条带,表明转化成功(图2A)。提取含有重组转化子的AH109菌总蛋白进行蛋白电泳,再用抗GAL4DNA-BD抗体检测诱饵蛋白的表达,结果显示在相对分子质量73×103处有BD-JNKK2融合蛋白的表达(图2B),表明含诱饵重组子的AH109酵母菌构建成功。
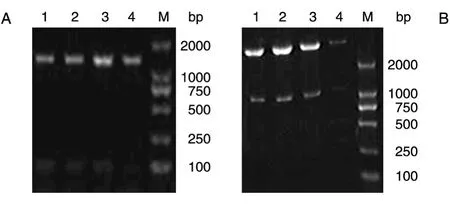
图1 诱饵载体pGBKT7-JNKK2的构建
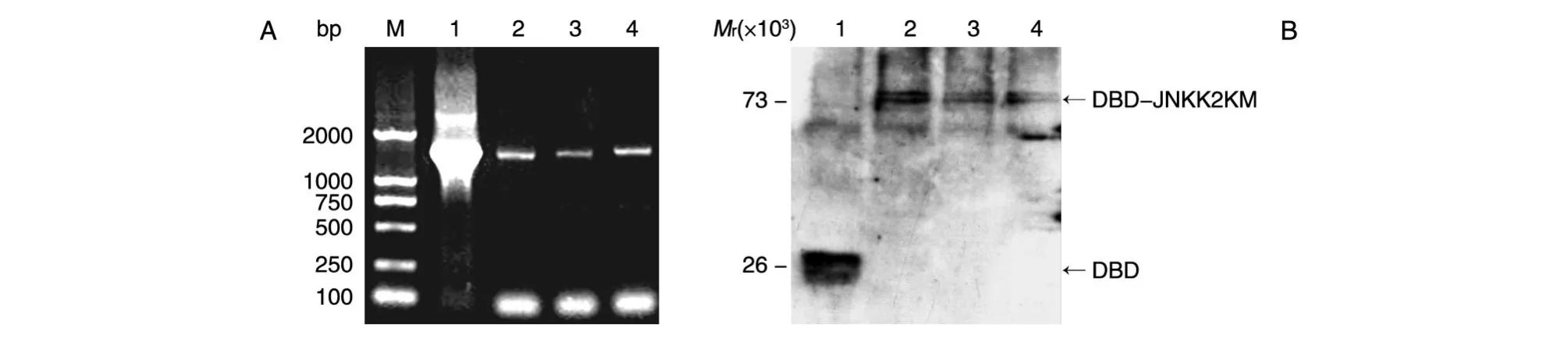
图2 诱饵蛋白的表达检测
2.3 重组诱饵表达质粒pGBKT7-JNKK2KM毒性与自我激活性检测
将上述重组成功的pGBKT7-JNKK2KM与空载体pGBKT7分别转入AH109酵母菌,观测其在SD/-Trp平板上生长状况。结果显示含诱饵质粒的AH109酵母菌在SD/-Trp上生长良好,二者生长速度相近、克隆大小相当,表明诱饵质粒对宿主酵母没有明显毒性(图3A)。同时自我激活实验显示AH109/pGBKT7-JNKK2KM在SD/-Ade/-His/-Trp/X-α-Gal平板上不能生长,也不能使X-α-Gal平板变蓝(图3B),排除了pGBKT7-JNKK2K具有自激活宿主菌AH109的作用。说明构建的诱饵表达载体pGBKT7-JNKK2KM可用于酵母双杂交系统,通过文库筛选相互作用蛋白。
2.4 cDNA文库交配效率计算
交配液涂板前,取出10μL按表1中比例稀释,取出100μL分别接种至SD/-Trp、SD/-Leu、SD/-Leu/-Trp平板,倒置培养克隆,生长计数见表1。已知2个交配伴侣的各自活力大小,较少的一方处于“限制性位置”。本实验中交配效率(二倍体数/限制方菌落数×100%=664/1469×100%=45.2%)远高于2%的最低筛选要求。
2.5 阳性克隆的分离与验证
AH109/pGBKT7-JNKK2KM与Y187文库酵母菌接合培养,在SD/-Ade/-His/-Leu/-Trp/平板上陆续收集>2mm的酵母菌落,共计120个。经SD/-Leu/-Trp板划线分离,挑取出3个克隆再涂在SD/-Ade/-His/-Leu/-Trp/X-α-Gal或 SD/-Ade/-His/-Leu/-Trp/H2O平板上进行验证,变蓝色为阳性克隆(图4A)。阳性克隆再次划线分离与验证2~3次,最后选取每次验证均变蓝色的克隆32个。提取酵母质粒,用pGADT7载体对应的常规AD引物做PCR鉴定,获得11个阳性克隆,后经将酵母质粒转化大肠杆菌DH5α后,提取大肠杆菌质粒,用HindⅢ酶切鉴定,结果显示11个克隆酶切结果均为阳性(图4B)。
2.6 阳性克隆基因序列的测定与分析
用pGADT7载体对应的常规AD测序引物[9]对阳性克隆进行测序,分析插入文库cDNA序列。序列结果用NCBI的BLAST在线比对工具进行比对。把测序结果中相位正确的克隆确认为真正的阳性克隆,最终确定11个阳性克隆中有3个为已知的编码基因(表2)。经序列比对发现,6号克隆与泛素样修饰物激活酶3(UBA3)编码区1335~1392位匹配,9号克隆与3号染色体开放读框60(ORF60)的CDS区完全匹配,25号克隆与鸟嘌呤核苷酸结合蛋白β多肽2样蛋白1(GNB2L1)编码区380~1060区匹配。以9号克隆质粒为模板调取ORF60全长编码序列,以25号克隆为模板调取GNB2L1C端681bp序列(GNB2L1C)。考虑到UBA3匹配序列较短,我们全合成UBA3C端1102~1392位291bp序列(UBA3C)。为便于研究以上调取的基因,我们在N端均加入Flag多肽标签,并克隆到真核表达载体pcDNA3.1(-)上。
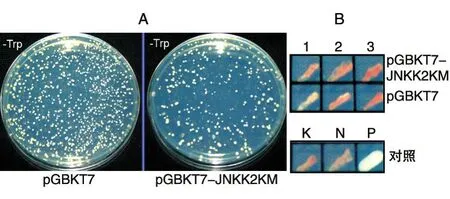
图3 诱饵蛋白的毒性与自激活性检测
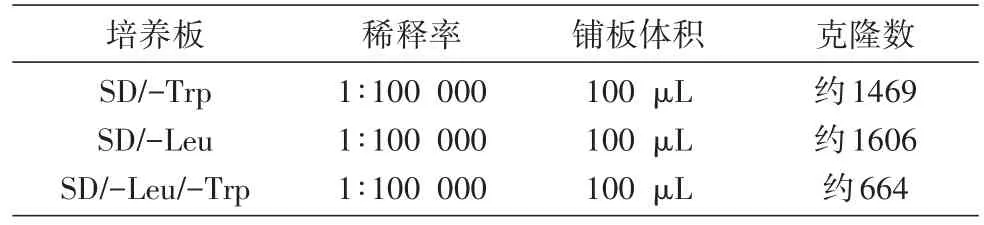
表1 交配液按比例稀释后在不同平板上克隆情况
2.7 免疫共沉淀验证阳性克隆与JNKK2蛋白的相互作用
分别转染 ORF60、GNB2L1C、UBA3C 质粒到293T细胞,同时共转GFP-JNKK2质粒,共培养24h后提取细胞蛋白,用Flag抗体M2免疫沉淀目的蛋白,用兔抗人JNKK2抗体检测相互作用条带。结果如图5所示,3个克隆均能不同程度地沉淀出GFP-JNKK2蛋白条带,而空载体对照则没有沉淀条带,表明克隆均能与JNKK2蛋白在哺乳动物细胞内相互结合。
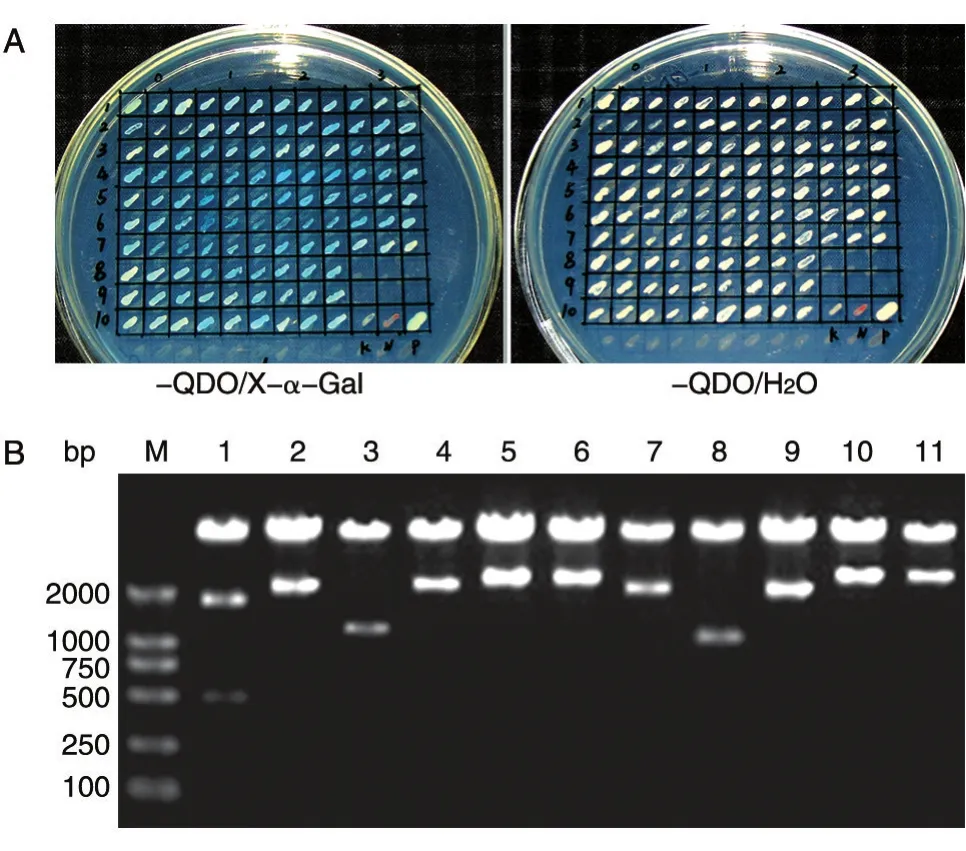
图4 初筛克隆的验证

表2 筛选文库阳性克隆编码基因信息
3 讨论
酵母双杂交系统是分析蛋白相互作用强有力的研究方法之一,作为筛选相互作用蛋白的工具得到了广泛应用[7-8]。本研究以JNKK2KM分子为“诱饵”,应用酵母双杂交系统对人胎肝文库进行筛选,初步筛选出120个克隆,后经多轮筛选,合并重复序列,最终筛选出11个阳性克隆。质粒测序后进行序列比对分析,得到3个编码基因ORF60、GNB2L1、UBA3。经免疫共沉淀实验验证,3种蛋白均可以与JNKK2蛋白发生相互作用。
ORF60属于线粒体复合物Ⅰ家族,相关研究较少,仅有的几篇报道表明其对线粒体组装至关重要;另有研究表明新生儿氧化磷酸化失能综合征(disordersoftheoxidativephosphorylation,OX⁃PHOS)与其功能突变密切相关[10]。ORF60与JNKK2相互作用的生物学意义仍待进一步挖掘。
GNB2L1属于Gβ样支架蛋白[11-12],相对分子质量约为36×103,其编码基因位于第5号染色体的q35.3区。GNB2L1能激活蛋白激酶C(PKC),又称为活化的蛋白激酶C受体1(receptorofacti⁃vatedC-kinase1,RACK1)。已发现能与GNB2L1结合的蛋白有近140种,其中包括许多重要的炎性信号转导分子,如 PKC、MKK7、Src、PDE4D5、PI3K等[12-14]。GNB2L1作为接头蛋白,参与调控细胞生长、凋亡、迁移、分化等过程,在肿瘤发生、发展中发挥重要作用[12-13]。人非小细胞肺癌中GNB2L1的mRNA水平比正常肺组织高4倍,在结肠癌中的水平比正常结肠组织高近20倍[15]。GNB2L1还在口腔鳞癌、乳腺癌和肺腺癌中高表达[16-18]。近期,顾建新课题组发现肝细胞癌组织中GNB2L1异常高表达,且GNB2L1与不良预后呈正相关[19]。提示GNB2L1很可能通过与JNKK2的相互作用来影响JNK活性,从而促进肿瘤发生。
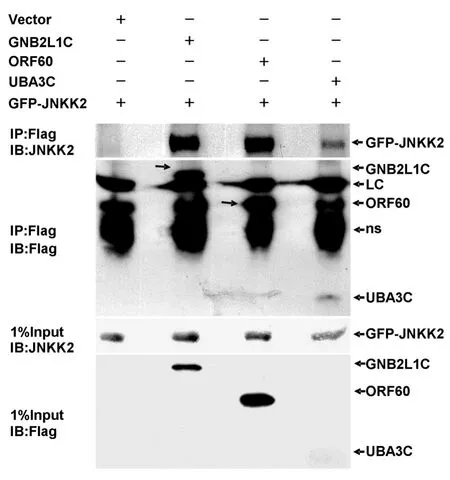
图5 免疫共沉淀验证阳性克隆与JNKK2蛋白的相互作用
UBA3是泛素化样修饰E1蛋白家族成员,通过结合β淀粉样蛋白前体蛋白结合蛋白(amyloidbeta precursorproteinbindingprotein,APPBP1)形成异二聚体,进而激活对泛素样蛋白NEDD8(neuralpre⁃cursorcell-expresseddevelopmentallydownregulated 8)的修饰作用,NEDD8修饰(neddylation)的功能主要体现在调节蛋白质之间的相互作用、调节转录因子的活性以及拮抗泛素化等。已有研究表明ned⁃dylation系统参与调控多种肿瘤的恶性进程:免疫组化显示肝内胆管癌样本中有2/3表现出neddylation E1酶(NAE1,UBA3)、E2酶(UBC12)及NEDD8高表达[20];人恶性胶质瘤中neddylation通路高度活化,与疾病的恶性程度正相关[21];人肺腺癌和鳞状细胞癌中neddylation通路同样高度活化[22]。Neddylation抑制剂MLN4924可以导致肿瘤细胞S期DNA合成紊乱,从而引起肿瘤细胞凋亡,具有很好的肿瘤治疗前景,目前已进入临床试验阶段[23]。以上提示,UBA3与JNKK2相互作用可能参与调控neddylation修饰过程,与肝癌的发生发展相关。
综上,我们通过酵母双杂交系统筛选出调控JNKK2-JNK通路的蛋白分子,针对所筛选的蛋白与JNKK2间相互作用,及该作用在肿瘤发生发展中所发挥的病理生理学意义的研究,具有很好的研究价值。后续希望能深入探讨JNKK2与新筛选蛋白间相互作用的病理生理学意义,更好地理解调控JNK-JNKK2通路的分子机制,为发现新的抗肝癌靶点奠定基础。
