抗结核药物靶点的研究进展
2018-06-13王霞朱宁屿姜威司书毅
王霞,朱宁屿,姜威,司书毅
抗结核药物靶点的研究进展
王霞,朱宁屿,姜威,司书毅
100050 北京,中国医学科学院北京协和医学院医药生物技术研究所国家新药(微生物)筛选实验室
结核病(tuberculosis,TB)是由结核分枝杆菌(mycobacterium tuberculosis,MTB)引起的一种慢性致死性疾病,目前是全世界十大致死疾病之一[1]。近些年来,由于抗结核药物的大量使用,抗结核作用位点的突变,艾滋病和结核耐药菌株的出现等原因,抗结核治疗面临严峻挑战。据世界卫生组织估算,2016 年,全球有 1040 万人患有结核病,170 万人因该病死亡(包括 40 万艾滋病毒感染者),有 60 万利福平耐药新发病例,其中有 49 万耐多药结核病患者[1]。耐多药结核病(MDR-TB)和广泛耐药结核病(XDR-TB)是控制和治疗结核病的重大挑战。然而,近几十年来仅有强生公司的新型结核分枝杆菌 ATP 合酶抑制剂贝达喹啉(bedaquiline)和日本大冢公司的德拉马尼(delamanid)两种抗结核药物被批准上市,而且目前已有耐药菌株出现及引起副作用的报道[2]。因此,寻找新的抗结核药物作用靶点,开发新型抗结核药物迫在眉睫。本文就近几年来有关抗结核药物靶点的研究作简要综述,旨在为研究新型抗结核药物以及发现潜在的新型抗结核药物靶点提供一些参考。
1 与 DNA/RNA 合成有关的药物靶点
1.1 DNA 回旋酶
DNA 回旋酶由两个亚蛋白单元 A(GyrA)和 B(GyrB)以异源四聚体的形式存在,DNA 回旋酶在人体内不存在,但却是细菌生存的必需物质,抑制其作用将导致细菌死亡[3]。
氧氟沙星、左氧氟沙星和莫西沙星等喹诺酮类抗结核药物主要作用于结核分枝杆菌 GyrA 蛋白,阻止 DNA 的复制和转录,从而杀死 MTB。但是,由于耐药性的出现,相继出现了一些以 GyrB 为靶点的药物,如苯并咪唑、尿素酶等[4]。Medapi 等[5]对已经报道的 GyrB 抑制剂进行结构优化,合成了 46 种新型喹啉衍生物。Locher 等[6]在最新研究中得到了一个 GyrB 抑制剂——VXc-486(图 1),该化合物对临床分离的结核分枝杆菌敏感株和耐药株的 MIC 分别为 0.03 ~ 0.30 μg/ml 和 0.08 ~ 5.48 μg/ml,可以在低氧环境下杀死细胞内潜伏的 MTB。通过对 VXc-486 化学结构进行改造,发现磷酸化的 VXc-486 比其本身抑菌效果更好,所以该化合物值得进行深入研究。
1.2 RNA 聚合酶
细菌的 RNA 聚合酶是一种由 α、β、β'、σ、ω 五种蛋白亚基组成的复合酶,每分子 RNA 聚合酶除有两个 α 亚基外,其余亚基均只有一个。利福平与 RNA 聚合酶的 β 亚单位结合,阻止 RNA 聚合酶与 DNA 连接,使 RNA 的转录和合成受阻,从而达到杀菌目的。利福平对快速增殖期和间断繁殖的 MTB 杀菌力都很强,与链霉素和异烟肼联用时可使它们的治疗效果增强。利福平耐药性的产生主要是由 RNA 聚合酶 β 亚基的基因突变所致。Hameed 等[7]发现531 基因突变与利福平耐药相关。Ullah 等[8]发现利福平耐药菌株的基因突变发生在长为 81 bp 的中心区域的 511、513、522、526 或 531 位置,这些位置的氨基酸可能是产生耐药性的关键氨基酸。
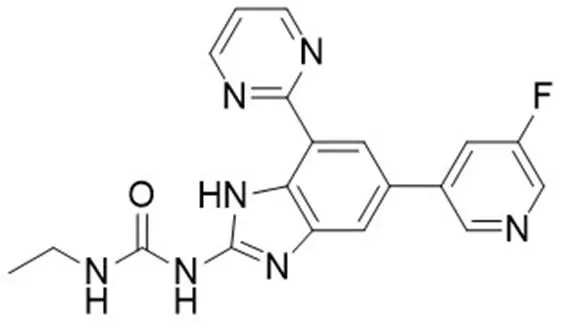
图 1 GyrB 抑制剂氨基苯并咪唑(VXc-486)
1.3 次黄嘌呤鸟嘌呤磷酸核糖转移酶
次黄嘌呤鸟嘌呤磷酸核糖转移酶(HGPRT)是一种细胞质酶,在体内广泛存在,它不仅参与嘌呤碱基的补救合成途径,对 DNA/RNA 的合成起重要作用,而且关系到嘌呤类药物的代谢,是调控该类药物药理效应和毒性反应的关键酶。Ducati 等[9]阐述了 MTB 嘌呤补救途径的酶作为开发新的抗分枝杆菌药物的优势。Eng 等[10]证明其新合成的开环膦酸核苷类药物(ANPs)是 MTB HGPRT 竞争性抑制剂,能够抑制 MTB 生长。这些化合物的 MIC 可低至 4.5 μmol/L,对哺乳动物细胞毒性较低。同时,确定了一些 HGPRT 的晶体结构,其中 3 个与新型 ANPs 复合,一个与鸟苷酸及焦磷酸复合。这些数据为 ANPs 作为结核分枝杆菌 HGPRT 选择性抑制剂和抗结核药物的进一步研究提供了理论依据。
1.4 胸苷酸激酶
抗病毒化疗往往依赖于核苷类似物,核苷类似物经胞内激酶磷酸化可作用于病毒聚合酶,进而阻碍病毒 DNA 合成,杀死病毒。相比之下,核苷类似物作为抗菌药物的研究较少,但 MTB 胸苷酸激酶(TMK)是 DNA 复制必不可少的酶,可以作为设计新的抑制剂的靶点[11]。Naik 等[12]应用高通量筛选和核磁共振的方法,发现了两类新型 TMK 抑制剂,分别是氰基吡啶酮类和萘啶类。以往的 TMK 抑制剂都是胸苷单磷酸类似物或者是含有胸苷单磷酸结构,而这两类化合物是非胸腺嘧啶核苷类 TMK 抑制剂,对 TMK 具有较好的选择性。
2 与细胞结构有关的药物靶点
2.1 与细胞脂类代谢相关的药物靶点
脂类是 MTB 细胞膜和细胞壁的主要成分,其代谢与 MTB 的生长繁殖密切相关。分枝菌酸是分枝杆菌细胞壁的一种组成成分,在分枝杆菌细胞壁结构和抗渗性方面起着至关重要的作用,是分枝杆菌毒力的关键因子。
2.1.1 烯酰基载体蛋白还原酶 烯酰基载体蛋白还原酶(InhA)是脂肪酸生物合成途径中的关键酶。InhA 是一线抗结核药物异烟肼的一个比较重要的作用靶点[13]。Šink等[14]以一个四环的噻二唑为母核设计合成了一系列新的三环 InhA 抑制剂,其中化合物 8d(图 2)是这些化合物中最有效的,在 THP-1 巨噬细胞内表现出较好的抗菌活性,在人微粒体中有较好的清除率。Campen 等[15]发现 InhA 的过表达可以增加耻垢分枝杆菌对异烟肼的抗性,然而对异烟肼的类似物 NSC27607、NSC33759 和 NSC40350 的抗性并没有增加,说明这些类似物并不是直接作用于该靶点,为后续以 InhA 依赖的抗性机制研究提供了基础。Elitas[16]利用微流控芯片和自动延时显微镜,采用单细胞分析法对异烟肼的杀菌机制提出了新见解,认为异烟肼耐药与活的非可培养的细胞亚群有关,为更好地理解细菌的耐药机制及设计抗生素治疗药物提供了理论基础。
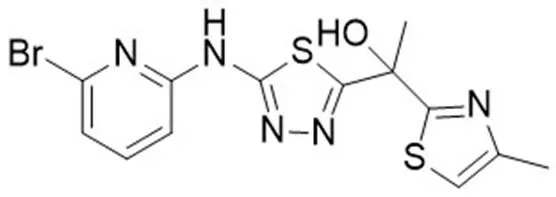
图 2 InhA 抑制剂 8d
2.1.2 β-酮酰-ACP 合成酶III β-酮酰-ACP 合成酶 III(FabH)催化脂肪酸生物合成的起始步骤,催化酰基辅酶 A 和丙二酰基载体蛋白(ACP)缩合,并可以受到终产物棕榈酰-ACP 的反馈抑制,使脂肪酸合成循环终止,在细胞壁分枝菌酸的合成中起着重要的作用[17]。FabH 普遍存在于病原体中,是其生存必需的酶,在人体中却无其同源蛋白,是一个潜在的抗结核药物靶点。从土壤诺卡菌属(sp.)中分离得到的天然产物 thiolactomycin(TLM)能够抑制 FabH 活性,设计合成的几个 TLM 衍生物在体外能更有效地抑制分枝菌酸的生物合成,且具有体内抗结核活性[18]。Brown 等[17]通过定点突变的方法研究影响 FabH 活性的关键氨基酸。结果显示,Arg46 是丙二酰单酰-AcpM 缩合作用的重要位点。His258 是水分子的一个定位点,可能促进 Cys122 的去质子化作用和转酰基作用。
2.1.3 泛酸合成酶 泛酸是 MTB 脂类物质合成必需的前体物质,且人体内不存在泛酸合成途径。MTB 泛酸合成途径主要有 4 个酶参与,其中泛酸合成酶(PanC)催化泛酸合成的最后一步反应,是关键的限速酶,所以 PanC 可以作为抗结核药物的靶点[19]。Zheng 等[20]证明 PanC 中高度保守的 His44、His47、Asn69、Gln72、Lys160 残基对泛酸合成酶反应中间体的结构稳定性是必需的。Velaparthi等[19]对一系列具有泛酸合成酶抑制作用的化合物进行研究,发现叔丁基和吡唑部分是PanC 抑制剂的关键药效团,吡唑基团的芳基部分具有良好的耐受性,这些研究结果对开发 PanC 抑制剂具有指导作用。
2.1.4 DesA3 DesA3(由基因编码)是一种膜结合硬脂酰辅酶 A Δ9脱氢酶,与氧化还原酶 Rv3230c(基因编码)共同参与代谢反应,产生油酸[21]。油酸是 MTB 膜磷脂和甘油三酯的重要组成部分,对细菌的生存具有重要作用。戊氧苯硫脲是个二线抗结核药物,该药物主要作用于 DesA3,可与异烟肼联合用于治疗耐多药结核病[22]。Chang 和 Fox[22]通过实验研究发现在耻垢分枝杆菌中过表达 C-末端融合 6 个组氨酸标签或 myc 标签 DesA3,比带有正常 C-末端的 DesA3 具有更高的催化活性和稳定性。深入研究 C-末端序列对 DesA3 的作用,发现带电性的侧链、大的非极性侧链或 C-末端最后两个位置的残基上没有侧链时,对稳定重组构建的带标签的 DesA3 结构最为重要,而这些侧链在倒数第三个残基上时对 DesA3 的结构影响较小[23]。这些研究成果为以后采用结构修饰的方法来抑制 DesA3 活性提供了参考。
2.2 与细胞糖类代谢相关的药物靶点
2.2.1 L-鼠李糖合成相关酶 L-鼠李糖连接细胞壁肽聚糖和阿拉伯半乳聚糖(AG)两种组分,通过抑制其合成,可以影响 MTB 细胞壁的正常合成及生存。由于人体内不存在 L-鼠李糖,所以L-鼠李糖合成中相关酶可以作为具有较好选择性的药物靶点[24]。
L-鼠李糖的合成主要涉及 4 种酶促反应,参与反应的 4 种酶分别为葡萄糖-1-磷酸脱氧胸苷转移酶(RmlA)、dTDP-D-葡萄糖-4,6-脱水酶(RmlB)、dTDP-4-酮基-6-脱氧葡萄糖差向异构酶(RmlC)和 dTDP-4-酮-鼠李糖还原酶(RmlD)。Ma 等[24]建立了一种可以同时筛选 RmlB、RmlC 和 RmlD 3 种酶抑制剂的微量滴定板法,得到了 4 个抑制结核分枝杆菌生长的化合物。Shi 等[25]测定了 RmlB 的动力学特性,包括 pH 值、温度、金属离子的影响,为 RmlB 抑制剂的研究提供了参考。Ren 等[26]利用类药性预测、药效团模型筛选、分子对接等虚拟筛选方法得到了 20 个具有良好的吸收、分布、代谢、排泄和毒性性质的 RmlB 和 RmlC 抑制剂,这些化合物值得进一步的构效关系研究及生物活性分析,以期得到理想的抗结核候选化合物。
2.2.2 阿拉伯糖基转移酶 一线抗结核药物乙胺丁醇通过作用于阿拉伯糖基转移酶,抑制阿拉伯半乳聚糖聚合,从而抑制 MTB 细胞壁的生物合成。乙胺丁醇耐药与阿拉伯糖基转移酶的编码基因 embCAB 操纵子(包括、和3 个基因)表达增高或突变有关,但主要是突变所致[27-28]。Brossier 等[27]对其研究的 71 种临床分离的乙胺丁醇耐药结核菌株进行基因分析,发现菌株中 98% 的基因突变发生在 embCAB 操纵子区域,其中 70% 的基因突变发生在基因 306、406 或 497 密码子,13% 的基因突变在这 3 个位点外的 296 ~ 426 密码子之间,15% 的突变发生在-基因间隔区。乙胺丁醇耐药与 MDR 产生密切相关,且对乙胺丁醇耐药的菌株大多也对异烟肼和利福平两种抗结核药物具有耐药性[28]。
2.3 2-C-甲基-D-赤藓糖醇磷酸途径相关酶
2-C-甲基-D-赤藓糖醇磷酸(MEP)途径能够合成异戊酰焦磷酸或其同分异构体二甲烯丙基焦磷酸,作为前体物质合成异戊二烯化合物,存在于许多细菌、藻类、植物和疟原虫,但不存在于哺乳动物细胞中,已成为发现新的抗疟药物和抗菌化合物的关注点[29]。在结核分枝杆菌中,MEP 途径参与 MTB 的细胞壁和 ATP 的合成,DXP 还原异构酶(IspC)和胞苷酰基转移酶(IspD)是 MEP 途径关键酶。Haymond 等[30]建立了一种高通量筛选 IspC 和 IspD 抑制剂的方法,并对市售的天然产物提取物 LOPAC1280 化合物库进行了筛选,确定了筛选方法的有效性。鲁众阳等[31]得到了 IspD 的有效抑制剂 IMB-4901(图 3),其酶抑制活性较好(IC50= 19.3 μg/ml),同时具有抗 MTB 活性(MIC = 1.6 μg/ml)。酶动力学研究表明,IMB-4901 是 IspD 反应底物三磷酸胞苷和 MEP 的竞争性抑制剂,成为以 IspD 为靶点的新型抗结核药物先导化合物。
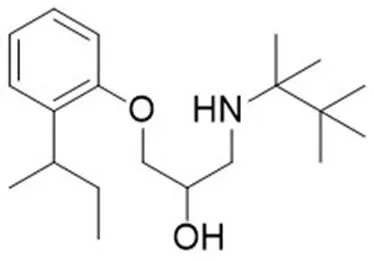
图 3 IspD 抑制剂 IMB-4901
2.4 HspX 蛋白
HspX 蛋白是 MTB 中通过基因编码的一个 16 kD 的 α-晶状体蛋白,该蛋白在 MTB 潜伏期时高度表达于细胞壁,这表明HspX 蛋白与 MTB 潜伏感染或持留性有关[32]。Hu 等[33]将缺陷株(ΔhspX)作为潜在的抑制模型,分别用 ΔhspX 和野生菌株感染正常 BALB/c 小鼠,发现感染 ΔhspX 的复发率明显低于感染野生菌株的复发率。Castro-Garza 等[34]分别用标准 ELISA 法和 Luminex xMAP®珠捕获 ELISA 法检测急性和潜伏性结核感染患者(潜伏结核感染为对先前获得的 MTB 抗原有持续免疫反应但没有临床证据证明表现为活动性 TB 的状态)血清中 HspX 蛋白及抗 HspX 的 IgG 和 IgM 抗体含量,结果显示潜伏性结核感染患者组比活动性结核病组表现出较高的 HspX 蛋白水平及抗体水平(= 0.003)。因此血清中 HspX 蛋白和抗 HspX IgM 抗体的存在可能是潜伏性结核病的标记物。
3 与必需的细胞功能有关的靶点
3.1 腺嘌呤核苷三磷酸合成酶
贝达喹啉(Bdq,图 4),曾被命名为 TMC207 或 R207910,是一种二芳基喹啉类化合物,被美国食品药品监督管理局(FDA)批准用于治疗耐多药肺结核病。Bdq 作用于ATP合成酶,导致 ATP 耗竭,从而引起 MTB 死亡。Bdq 对快速生长期、休眠期及具有耐药性的 MTB 都有抑制活性,且对 MTB 有高度选择性,对其他常见的普通细菌几乎无活性[35]。Bdq 耐药性的产生与 MTB基因和基因有关:基因 63 位密码子的基因突变使 Bdq 与 ATP 合酶 C 亚基结合受阻;基因 63 位或 66 位密码子的基因突变将阻止 Bdq 进入 ATP 合酶的结合口袋;基因突变会产生 Bdq 与氯法齐明交叉耐药[36-37]。Bdq 可能影响患者的心电活动,导致患者心脏节律异常,出现恶心、关节痛和头痛等不良反应,其长期疗效和安全性的关键数据是不完整的,所以应该慎用 Bdq[38]。
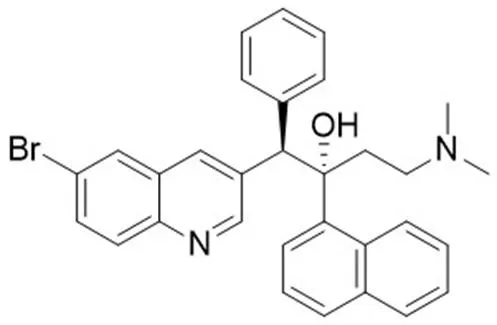
图 4 ATP 合成酶抑制剂贝达喹啉
3.2 Ser/Thr 蛋白激酶 G
MTB 共包含两组信号转导系统,双组分系统和 Ser/Thr 蛋白激酶信号系统。MTB 共编码 11 种 Ser/Thr 蛋白激酶,分别为 PknA、PknB、PknD、PknE、PknF、PknG、PknH、PknI、PknJ、PknK 和 PknL。这些激酶主要参与菌体的生长发育、毒力以及能量代谢等生理过程[39]。其中,PknA、PknB 和 PknG 3 种蛋白激酶对 MTB 的体外生长很重要。深入研究发现,敲除基因的突变株侵染巨噬细胞时,被溶酶体消化的巨噬细胞数量明显多于 PknG 功能正常的菌株;在耻垢分枝杆菌中表达 PknG 也可以显著地阻断吞噬体与溶酶体的融合[40]。最新研究表明,PknG 可能通过调节代谢途径中 GarA 的磷酸化状态,阻断溶酶体降解巨噬细胞内的分枝杆菌,从而维持分枝杆菌在细胞内的生存[41]。Singh 等[42]找到了 3 个具有明显 PknG抑制活性的化合物。通过卡介苗感染 THP-1 巨噬细胞,发现化合物 NRB04248(图 5)能够抑制卡介菌的增长且对宿主的巨噬细胞无毒。上述研究结果表明,化合物 NRB04248 可以作为 PknG 抑制剂进行深入研究。
3.3 丝状温敏蛋白 Z
丝状温敏蛋白Z(FtsZ)是一种 GTP 酶,普遍分布在细菌体内,参与细菌的有丝分裂过程。当 FtsZ 与 GTP 结合时,FtsZ 自我聚合成多聚体,随后在菌体膜中心形成 Z-ring。在其他分裂的蛋白质的帮助下,Z-ring 逐渐缩小形成隔膜,使菌体一分为二。当 GTP 水解后,FtsZ 聚合体又会再度分离成单体。若阻断 FtsZ 蛋白的 GTP 酶活性,细菌分裂会受到抑制,从而使细菌死亡[43]。由于 FtsZ 是哺乳动物微管蛋白 tubulin 的对等蛋白,所以已知的微管蛋白抑制剂可作为开发 FtsZ 抑制剂的一个很好的起点。Park 等[44]分析先前报道的 tubulin 抑制剂,将苯并咪唑作为母体,合成了 376 个三取代的苯并咪唑衍生物,并对其生物活性进行研究,最终得到了一个 MIC = 0.63 μg/ml,与MTB-FtsZ 亲和力为 1.32 μmol/L 的化合物。林媛等[45]建立了以 FtsZ 为靶点的高通量筛选模型,利用该模型筛选得到的化合物 212H(图 6),212H 可以抑制 FtsZ 的 GTP 酶活性,并且可以抑制 FtsZ 的聚合。以 FtsZ 为靶点,通过虚拟筛选得到的化合物 228C、144G和 90D(图 6)均能够抑制 FtsZ 的 GTP 酶活性,均具有较好的抗耻垢分枝杆菌活性,为以 FtsZ 为靶点的抗结核药物研究提供了较好的分子探针[46]。
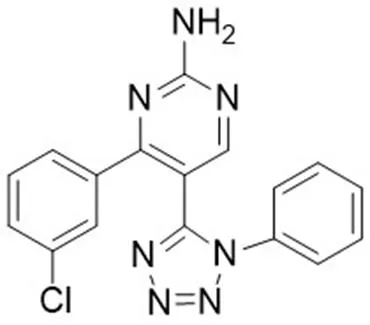
图 5 PknG 抑制剂 NRB04248
3.4 与莽草酸途径有关的靶点
莽草酸途径是细菌、真菌、藻类和高等植物合成芳香族氨基酸、叶酸、分枝菌酸、泛醌、萘醌等必需物质的一条重要代谢途径。莽草酸途径只存在于细菌、真菌、藻类和植物中,人体内不存在该途径,可以此途径中的关键酶作为抗结核靶点[47]。
近年来,对催化莽草酸途径第五步反应的莽草酸激酶(SK)研究较多。Pereira 等[48]对 SK 与 MgADP 和莽草酸的三维结构进行分析,发现莽草酸的羧基主要与 SK 的 Arg58、Gly81 和 Arg136 相互作用,莽草酸的羟基主要与 SK 的 Asp34 和 Gly80 相互作用。Pereira 等[49]进一步对SK 作为抗结核药物靶点进行了总结,为合理设计抗结核药物提供了方向。Blanco 等[50]设计了一系列莽草酸分子类似物,并使用分子动力学模拟方法深入研究了这一系列底物类似物与 MtSK 蛋白的结合方式。虽然现在对 SK 的研究大多数停留在通过计算机辅助药物设计,对天然底物进行结构改造指导合成底物类似物阶段,但合成的化合物与 SK 的亲和力往往较好,所以进一步开展 SK 作为抗结核药物靶点的研究是有价值的。
3.5 与铁载体合成相关的靶点
MTB 必须合成分泌铁载体来转运铁,铁在 MTB 许多重要的生物学过程中起辅助作用,有效的铁摄取对 MTB 生存至关重要。铁载体合成过程中的关键酶包括 MbtA、MbtB 和调节蛋白 IdeR,其中对 MbtA 抑制剂的研究较多。Labello 等[51]获得了亲和力跨 6 个数量级的 31 个抑制剂,通过研究 MbtA 与抑制剂之间的构效关系,预测了MbtA 第二代抑制剂。Maganti 等[52]通过分子动力学模拟和分子对接的方法探索 MbtA 的蛋白结构,为作为抗结核治疗的 MbtA 抑制剂的合理设计提供了参考。Meneghetti等[53]证明了 MbtA 抑制剂在小鼠体内的抗结核作用,提示靶向铁摄取系统可以作为一种有效的抗结核策略。
3.6 P450酶系
许多细菌 P450酶系的酶不超过 5 种,大肠杆菌中甚至没有 P450酶系,而在 MTB 中 P450酶系却含有 20 多种酶,表明其在 MTB 代谢中发挥着重要的作用,也表明其中的一些酶很有可能成为新的抗结核药物靶点[54]。近年来,对MTB 中 P450酶系 CYP51、CYP121和 CYP1413 种酶的研究较多。McLean 等[55]研究发现,唑类药物对 CYP121的亲和力比 CYP51高,而且一系列唑类药物对 CYP121的亲和力大小与其潜在的抗结核活性相关,说明 CYP121可能才是唑类药物的真正靶点。对 CYP51和 CYP121的晶体结构进行研究,探索两种酶的生物学功能和底物选择性,为设计以 P450氧化还原系统为靶点的抗结核药物提供基础[54, 56]。Salehi 等[57]对 MTB 基因组中可能存在免疫原性的重组蛋白进行了免疫原性评价,发现 P450酶系 CYP141蛋白可以作为 MTB 候选疫苗进行深入研究。
4 小结
除上述抑制结核分枝杆菌 DNA 合成、RNA 合成、细胞壁合成及能量代谢途径的靶点外,与细菌蛋白质合成有关的肽脱甲酰基酶、与细胞壁合成相关的肌醇-1-磷酸合成酶、与电子传递链有关的 1,4-二羟基-2-萘甲酰辅酶 A 合成酶以及与 DNA 复制和修复机制相关的酶和蛋白质等也可以作为抗结核药物的潜在靶点。随着生物技术的发展,通过基因组测序及基因敲除等技术比较 MTB 基因组和人类基因组差异,可在 MTB 各种代谢途径中寻找理想的抗结核药物靶点。总之,不管靶向何种实体,对结核分枝杆菌的生存和致病途径进行多个方向的思考和研究将有助于人们开发抗结核药物,以实现对结核病的控制和治疗。
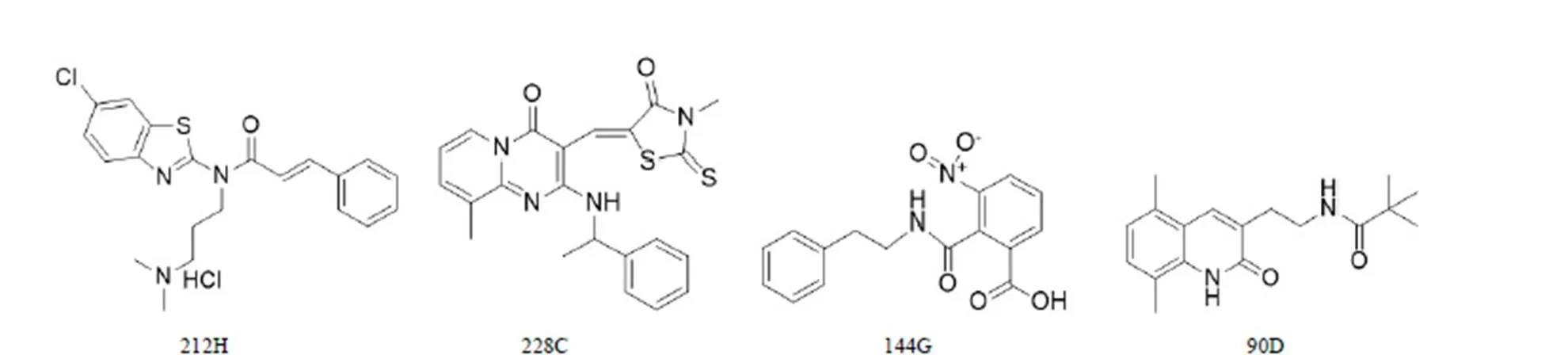
图 6 FtsZ 抑制剂
[1] World Health Organization. Global tuberculosis report 2017, Geneva: WHO Press, 2017:1-10.
[2] Bloemberg GV, Keller PM, Stucki D, et al. Acquired resistance to bedaquiline and delamanid in therapy for tuberculosis. N Engl J Med, 2015, 373(20):1986-1988.
[3] Mdluli K, Ma Z. Mycobacterium tuberculosis DNA gyrase as a target for drug discovery. Infect Disord Drug Targets, 2007, 7(2):159-168.
[4] Chaudhari K, Surana S, Jain P, et al. Mycobacterium tuberculosis (MTB) GyrB inhibitors: An attractive approach for developing novel drugs against TB. Eur J Med Chem, 2016, 124:160-185.
[5] Medapi B, Suryadevara P, Renuka J, et al. 4-Aminoquinoline derivatives as novel Mycobacterium tuberculosis GyrB inhibitors: Structural optimization, synthesis and biological evaluation. EurJ Med Chem, 2015, 103:1-16.
[6] Locher CP, Jones SM, Hanzelka BL, et al. A novel inhibitor of gyrase B is a potent drug candidate for treatment of tuberculosis and nontuberculosis mycobacterial infections. Antimicrob Agents Chemother, 2015, 59(3):1455-1465.
[7] Hameed S, Moganeradj K, Mahmood N, et al. Sequence analysis of the rifampicin resistance determining region (RRDR) of rpoB gene in multidrug resistance confirmed and newly diagnosed tuberculosis patients of Punjab, Pakistan. PLoS One, 2017, 12(8):e0183363.
[8] Ullah I, Shah AA, Basit A, et al. Rifampicin resistance mutations in the 81 bp RRDR of rpoB gene in Mycobacterium tuberculosis clinical isolates using Xpert MTB/RIF in Khyber Pakhtunkhwa, Pakistan: a retrospective study. BMC Infect Dis, 2016, 16:413.
[9] Ducati RG, Breda A, Basso LA, et al. Purine salvage pathway in Mycobacterium tuberculosis. Curr Med Chem, 2011, 18(9):1258- 1275.
[10] Eng WS, Hockova D, Spacek P, et al. First crystal structures of Mycobacterium tuberculosis 6-oxopurine phosphoribosyltransferase: Complexes with GMP and pyrophosphate and with acyclic nucleoside phosphonates whose prodrugs have antituberculosis activity. J Med Chem, 2015, 58(11):4822-4838.
[11] Van Calenbergh S, Pochet S, Munier-Lehmann H. Drug design and identification of potent leads against mycobacterium tuberculosis thymidine monophosphate kinase. Curr Top Med Chem, 2012, 12(7): 694-705.
[12] Naik M, Raichurkar A, Bandodkar BS, et al. Structure guided lead generation for M. tuberculosis thymidylate kinase (Mtb TMK): discovery of 3-cyanopyridone and 1,6-naphthyridin-2-one as potent inhibitors. J Med Chem, 2015, 58(2):753-766.
[13] Tseng ST, Tai CH, Li CR, et al. The mutations of katG and inhA genes of isoniazid-resistant Mycobacterium tuberculosis isolates in Taiwan. J Microbiol Immunol Infect, 2015, 48(3):249-255.
[14] Šink R, Sosič I, Živec M, et al. Design, synthesis, and evaluation of new thiadiazole-based direct inhibitors of enoyl acyl carrier protein reductase (InhA) for the treatment of tuberculosis. J Med Chem, 2015, 58(2):613-624.
[15] Campen RL, Ackerley DF, Cook GM, et al.Development of a Mycobacterium smegmatis transposon mutant array for characterising the mechanism of action of tuberculosis drugs: Findings with isoniazid and its structural analogues. Tuberculosis (Edinb), 2015, 95(4):432-439.
[16] Elitas M. Isoniazid killing of Mycobacterium smegmatis NADH pyrophosphatase mutant at single-cell level using microfluidics and time-lapse microscopy. Sci Rep, 2017, 7(1):10770.
[17] Brown AK, Sridharan S, Kremer L, et al. Probing the mechanism of the Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein synthase III mtFabH: factors influencing catalysis and substrate specificity. J Biol Chem, 2005, 280(37):32539-32547.
[18] Kremer L, Douglas JD, Baulard AR, et al. Thiolactomycin and related analogues as novel anti-mycobacterial agents targeting KasA and KasB condensing enzymes in Mycobacterium tuberculosis. J Biol Chem, 2000, 275(22):16857-16864.
[19] Velaparthi S, Brunsteiner M, Uddin R, et al. 5-tert-butyl-N-pyrazol-4- yl-4,5,6,7-tetrahydrobenzo[d]isoxazole-3-carboxamide derivatives as novel potent inhibitors of Mycobacterium tuberculosis pantothenate synthetase: initiating a quest for new antitubercular drugs. J Med Chem, 2008, 51(7):1999-2002.
[20] Zheng R, Dam TK, Brewer CF, et al. Active site residues in Mycobacterium tuberculosis pantothenate synthetase required in the formation and stabilization of the adenylate intermediate. Biochemistry, 2004, 43(22):7171-7178.
[21] Phetsuksiri B, Jackson M, Scherman H, et al. Unique mechanism of action of the thiourea drug isoxyl on Mycobacterium tuberculosis.J Biol Chem, 2003, 278(52):53123-53130.
[22] Chang Y, Fox BG. Identification of Rv3230c as the NADPH oxidoreductase of a two-protein DesA3 acyl-CoA desaturase in Mycobacterium tuberculosis H37Rv. Biochemistry, 2006, 45(45): 13476-13486.
[23] Chang Y, Wesenberg GE, Bingman CA, et al. In vivo inactivation of the mycobacterial integral membrane stearoyl coenzyme A desaturase DesA3 by a C-terminus-specific degradation process. J Bacteriol, 2008, 190(20):6686-6696.
[24] Ma Y, Stern RJ, Scherman MS, et al. Drug targeting Mycobacterium tuberculosis cell wall synthesis: genetics of dTDP-rhamnose synthetic enzymes and development of a microtiter plate-based screen for inhibitors of conversion of dTDP-glucose to dTDP-rhamnose. Antimicrob Agents Chemother, 2001, 45(5):1407-1416.
[25] Shi X, Sha S, Liu L, et al. A 96-well microtiter plate assay for high-throughput screening of Mycobacterium tuberculosis dTDP-d-glucose 4,6-dehydratase inhibitors. Anal Biochem, 2016, 498:53-58.
[26] Ren JX, Qian HL, Huang YX, et al. Virtual screening for the identification of novel inhibitors of Mycobacterium tuberculosis cell wall synthesis: inhibitors targeting RmlB and RmlC. Comput Biol Med, 2015, 58:110-117.
[27] Brossier F, Sougakoff W, Bernard C, et al. Molecular analysis of the embCAB locus and embR gene involved in ethambutol resistance in clinical isolates of Mycobacterium tuberculosis in France. Antimicrob Agents Chemother, 2015, 59(8):4800-4808.
[28] Margaryan H, Rusch-Gerdes S, Hayrapetyan A, et al. Ethambutol-resistance testing by mutation detection using MTBDRsl. Int J Mycobacteriol, 2016, 5 Suppl 1:S50.
[29] Saggu GS, Pala ZR, Garg S, et al. New insight into isoprenoids biosynthesis process and future prospects for drug designing in plasmodium. Front Microbiol, 2016, 7:1421.
[30] Haymond A, Dowdy T, Johny C, et al. A high-throughput screening campaign to identify inhibitors of DXP reductoisomerase (IspC) and MEP cytidylyltransferase (IspD). Anal Biochem, 2017, 542:63-75.
[31] Lu ZY, Yang YH, Meng JZ, et al. Screening of novel antitubercular drugs targeting IspD. Chin J New Drugs, 2015, 24(17):1947-1953. (in Chinese)鲁众阳, 杨延辉, 蒙建州, 等. 以IspD为靶点的新型抗结核药物筛选研究. 中国新药杂志, 2015, 24(17):1947-1953.
[32] Hu Y, Coates AR. Transcription of the stationary-phase-associated hspX gene of Mycobacterium tuberculosis is inversely related to synthesis of the 16-kilodalton protein. J Bacteriol, 1999, 181(5):1380- 1387.
[33] Hu Y, Liu A, Menendez MC, et al. HspX knock-out in Mycobacterium tuberculosis leads to shorter antibiotic treatment and lower relapse rate in a mouse model--a potential novel therapeutic target. Tuberculosis (Edinb), 2015, 95(1):31-36.
[34] Castro-Garza J, Garcia-Jacobo P, Rivera-Morales LG, et al. Detection of anti-HspX antibodies and HspX protein in patient sera for the identification of recent latent infection by Mycobacterium tuberculosis. PLoS One, 2017, 12(8):e0181714.
[35] Yadav S, Rawal G, Baxi M. Bedaquiline: a novel antitubercular agent for the treatment of multidrug-resistant tuberculosis. J Clin Diagn Res, 2016, 10(8):FM01-FM02.
[36] Petrella S, Cambau E, Chauffour A, et al. Genetic basis for natural and acquired resistance to the diarylquinoline R207910 in mycobacteria. Antimicrob Agents Chemother, 2006, 50(8):2853-2856.
[37] Huitric E, Verhasselt P, Koul A, et al. Rates and mechanisms of resistance development in Mycobacterium tuberculosis to a novel diarylquinoline ATP synthase inhibitor. Antimicrob Agents Chemother, 2010, 54(3):1022-1028.
[38] Nguyen TV, Cao TB, Akkerman OW, et al. Bedaquiline as part of combination therapy in adults with pulmonary multi-drug resistant tuberculosis. Expert Rev Clin Pharmacol, 2016, 9(8):1025-1037.
[39] Cole ST, Brosch R, Parkhill J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature, 1998, 393(6685):537-544.
[40] Walburger A, Koul A, Ferrari G, et al. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science, 2004, 304(5678):1800-1804.
[41] Rieck B, Degiacomi G, Zimmermann M, et al. PknG senses amino acid availability to control metabolism and virulence of Mycobacterium tuberculosis. PLoS Pathog, 2017, 13(5):e1006399.
[42] Singh N, Tiwari S, Srivastava KK, et al. Identification of novel inhibitors of Mycobacterium tuberculosis PknG using pharmacophore based virtual screening, docking, molecular dynamics simulation, and their biological evaluation. J Chem Inf Model, 2015, 55(6):1120-1129.
[43] Löwe J, Amos LA. Crystal structure of the bacterial cell-division protein FtsZ. Nature, 1998, 391(6663):203-206.
[44] Park B, Awasthi D, Chowdhury SR, et al. Design, synthesis and evaluation of novel 2,5,6-trisubstituted benzimidazoles targeting FtsZas antitubercular agents. Bioorg Med Chem, 2014, 22(9):2602-2612.
[45] Lin Y, Zhu NY, Jiang JD, et al. The establishment and application of a high-throughput screening (HTS) assay for inhibitors of Mycobacterium tuberculosis FtsZ. Chin J Antibiot, 2015, 40(3):166- 170, 212. (in Chinese)林媛, 朱宁屿, 蒋建东, 等. 以FtsZ为靶点的抗结核药物筛选模型的建立及应用. 中国抗生素杂志, 2015, 40(3):166-170, 212.
[46] Lin Y, Zhu NY, Jiang JD, et al. Screening of FtsZ-targeted compounds and their anti-tuberculosis activities.Chin J New Drugs, 2015, 24(5): 592-594, 600. (in Chinese)林媛, 朱宁屿, 蒋建东, 等. 靶向FtsZ先导化合物的筛选及其抗结核作用探讨. 中国新药杂志, 2015, 24(5):592-594, 600.
[47] Ducati RG, Basso LA, Santos DS. Mycobacterial shikimate pathway enzymes as targets for drug design. Curr Drug Targets, 2007, 8(3): 423-435.
[48] Pereira JH, de Oliveira JS, Canduri F, et al. Structure of shikimate kinase from Mycobacterium tuberculosis reveals the binding of shikimic acid. Acta Crystallogr D Biol Crystallogr, 2004, 60(Pt 12 Pt 2): 2310-2319.
[49] Pereira JH, Vasconcelos IB, Oliveira JS, et al. Shikimate kinase: a potential target for development of novel antitubercular agents. Curr Drug Targets, 2007, 8(3):459-468.
[50] Blanco B, Prado V, Lence E, et al. Mycobacterium tuberculosis shikimate kinase inhibitors: design and simulation studies of the catalytic turnover. J Am Chem Soc, 2013, 135(33):12366-12376.
[51] Labello NP, Bennett EM, Ferguson DM, et al. Quantitative three dimensional structure linear interaction energy model of 5'-O-[N-(salicyl)sulfamoyl]adenosine and the aryl acid adenylating enzyme MbtA. J Med Chem, 2008, 51(22):7154-7160.
[52] Maganti L, Open Source Drug Discovery C, Ghoshal N. Probing the structure of Mycobacterium tuberculosis MbtA: model validation using molecular dynamics simulations and docking studies. J Biomol Struct Dyn, 2014, 32(2):273-288.
[53] Meneghetti F, Villa S, Gelain A, et al. Iron acquisition pathways as targets for antitubercular drugs. Curr Med Chem, 2016, 23(35):4009- 4026.
[54] Munro AW, McLean KJ, Marshall KR, et al. Cytochromes P450: novel drug targets in the war against multidrug-resistant Mycobacterium tuberculosis. Biochem Soc Trans, 2003, 31(Pt 3):625- 630.
[55] McLean KJ, Marshall KR, Richmond A, et al. Azole antifungals are potent inhibitors of cytochrome P450 mono-oxygenases and bacterial growth in mycobacteria and streptomycetes. Microbiology, 2002, 148(Pt 10):2937-2949.
[56] McLean KJ, Belcher J, Driscoll MD, et al. The Mycobacterium tuberculosis cytochromes P450: physiology, biochemistry & molecular intervention. Future Med Chem, 2010, 2(8):1339-1353.
[57] Salehi M, Abdizadeh R, Pourgheysari B, et al. Evaluation of cellular immunogenicity of recombinant cytochrome p450 cyp141 protein of Mycobacterium tuberculosis in human and mouse model. Biologicals, 2018, pii:S1045-S1056(18)30022-8.
国家自然科学基金(81573474);北京协和医学院小规模特色办学专项青年教师培养项目(2015zlgc0745)
姜威,Email:imbjiangv@163.com;司书毅,Email:sisyimb@hotmail.com
2018-03-07
10.3969/j.issn.1673-713X.2018.03.011
