U-2.5% Nb合金表面的氢蚀特性
2018-06-12,,,,,
,, ,, ,
(1. 表面物理与化学重点实验室,绵阳 621908; 2. 中国工程物理研究院,绵阳 621900)
金属铀及其合金因具有良好的力学性能及核性能而作为重要的核材料在核工程中受到了广泛的应用。然而铀的化学性质非常活泼,极易与环境中的氧气、水蒸气[1-3]等相互作用而发生氧化腐蚀或氢化腐蚀。尤其是在无氧或少氧的潮湿环境中[4-5],铀与水蒸气反应生成的氢气会反过来作用于铀。铀与氢气反应生成的氢化物UH3极易被氧化,从而释放大量热,此时若产物突然暴露于空气中就会引起自燃,这给铀的长期储存及维护带来了严重的安全隐患。此外,氢蚀会严重降低铀材料的力学性能和结构完整性从而影响其使用性能[6],因此,深入研究铀及其合金氢蚀的特性至关重要。
目前国内外对于铀氢反应的动力学行为研究已经比较深入[7-9],而关于铀氢反应初期的成核机制却不明确。有研究表明铀氢反应会优先在晶界[10]、夹杂[11-12]、尖角[13]等缺陷处形核。近几年,有研究表明铀氢蚀的优先形核位点与金属自身的相组织结构也有关联:SHI等[14]的研究表明对于U-0.79% Ti合金,铀氢反应优先在α相形核;纪和菲等[15]的研究表明对于时效后发生了相分解的双相U-5.7% Nb合金,氢蚀会优先在相分解形成的贫铌α相上发生。
U-2.5% Nb合金是典型的双相组织[16],其抗氧化性能较好,抗氢蚀性能却较差。有研究表明[17],相较于单相纯U,U-2.5% Nb合金更易发生氢蚀,孕育期更短,反应速率更高,究其原因,国内外文献却鲜有提及。本工作利用加速腐蚀系统获得U-2.5% Nb合金氢化后的试样,通过表征其腐蚀特征,研究U-2.5% Nb合金氢蚀与组织结构之间的关联,从而进一步探讨其氢蚀特性。
1 试验
1.1 试样
试样采用U-2.5% Nb合金。其加工工艺为:铸造后控制冷却速率缓冷,在一定温度下退火,结构为双相组织α+γ1-2。试样尺寸为8 mm×4 mm×2 mm,用SiC砂纸(100~2 000号)逐级打磨试样所有表面,抛光后放入丙酮中超声清洗5 min,取出吹干后立即放入反应容器中快速密封进行氢化腐蚀试验,期间尽量缩短试样与空气接触的时间以尽量减少氧化膜的影响。
1.2 试验方法
氢化试验在图1所示的氢化腐蚀装置中进行。将试样密封于可视反应容器中,通过KH-7700型体视显微镜在线观察试样在反应容器内的反应情况,并以CCD(电荷耦合器件)相机每隔5 s采集一次图像,记录氢化反应过程。电加热炉套在反应容器外侧,反应温度由程序升温仪控制。压力由数字式高精度压力传感器测得,并输入电脑储存。试验前对整个系统抽真空至小于1 Pa,放入试样并确保整个系统的密封性,进一步抽真空至反应容器内压力也小于1 Pa,升温至160 ℃保温30 min以除去反应容器内壁及试样表面吸附的气氛,降温至反应温度(70 ℃),待温度稳定后通入一个大气压的氢气开始反应,其中氢气纯度为99.999%(体积分数)。在反应过程中,反应容器的体积与反应温度不变,根据理想气体状态方程(式1),通过检测氢压的降低来表征氢气的消耗量。当氢压下降3%时抽走氢气停止反应,获得氢化后试样。
PV=nRT
(1)
式中:P为气体压强, Pa;V为气体体积,m3;n为气体的物质的量,mol;T为体系温度,K;R是气体常量(比例常数),8.314 J/(mol·K)。
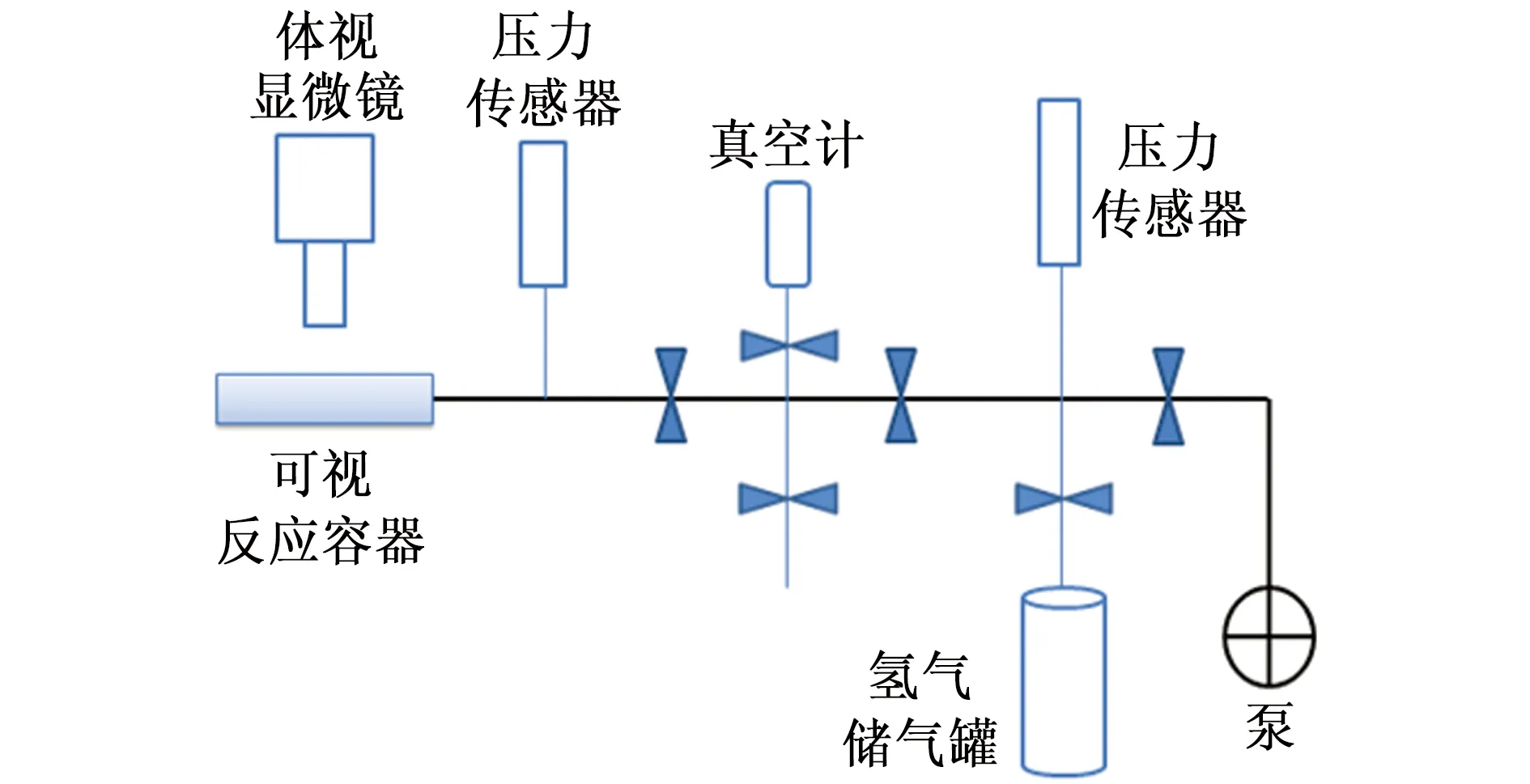
图1 氢化腐蚀装置示意图Fig. 1 The schematic of hydrogenation corrosion system
采用Empyream型X射线衍射(XRD)对氢化前后试样的成分进行分析,Cu靶(Kα),扫描步长为0.026°/步。采用HELIOS 600I型扫描电镜(SEM)和Lext OLS-4000型激光共聚焦扫描显微镜(LSCM)对氢化前后试样的形貌进行观察,从而进一步讨论U-2.5% Nb合金氢化的特性及作用机理。
2 结果与讨论
2.1 氢化前试样
由图2可见:对于U-2.5% Nb合金,当γ1相在固溶区冷却时,首先析出β相,664 ℃时发生共析反应,β相分解成α相和γ1相,在664~647 ℃冷却时,α相的铌含量有轻微的减小,但α相的量逐渐增加;同时,γ1相减小而铌含量增加。降温至647 ℃,γ1相发生偏共析反应生成 α相和γ2相,直至冷却至室温,合金为α相+γ2相的双相合金。然而由于本工作中试样是在非平衡的缓慢冷却条件下制得的,因而试样是由α相和成分介于γ1和γ2之间的γ1-2相组成的。由图3可见:U-2.5% Nb合金的结构为典型的珠光体层片状组织,这主要是由于试样是在缓慢冷却条件下制得的。XRD分析结果进一步明确了U-2.5% Nb合金的相组成为α相+γ1-2相。图4中可以看到明显的α相峰和相对较弱的γ1-2峰,与之相比,U-5.7% Nb合金[18]的γ1-2峰则强很多,这主要是由铌含量及热处理条件的不同造成的。
U-2.5% Nb合金为典型的珠光体片状结构,对其成分进行能谱分析,共测定了4个点,其中点1、2为较窄的深色区域,点3、4为较宽的浅色区域,见图5,所测结果见表1。由表1可见: 1、2点处的Nb含量远高于3、4点处的。这表明颜色较浅且较宽(约为1 μm)的区域为贫铌的α相,而相间于α相之间的颜色较深且较窄的区域为富铌的γ1-2相,即U-2.5% Nb合金是由较宽的贫铌α相和相间于其中的较窄区域的富铌γ1-2相组成的。
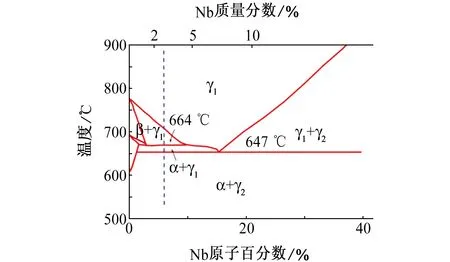
图2 U-Nb合金相图(富铌部分)[18]Fig. 2 Equilibrium phase diagram of U-Nb alloys(Nb enriched area)

图3 氢化前U-2.5% Nb合金的组织形貌Fig. 3 Structure morphology of U-2.5% Nb alloy before hydriding

图4 氢化前U-2.5% Nb合金的XRD结果Fig. 4 XRD result of the U-2.5% Nb alloy before hydriding

图5 U-2.5% Nb合金氢化前SEM形貌Fig. 5 SEM morphology of the U-2.5% Nb alloy before hydriding
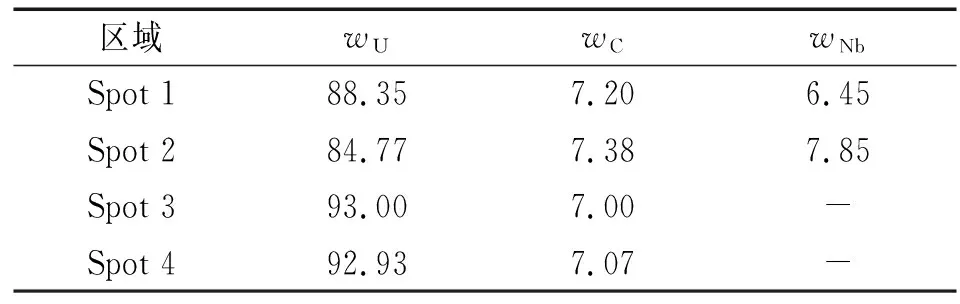
区域wUwCwNbSpot 188.357.206.45Spot 284.777.387.85Spot 393.007.00-Spot 492.937.07-
2.2 氢化后试样
早在20世纪60年代,就有研究表明铀的氢化腐蚀是不连续的点蚀,且在空间上随机分布[19-20]。试验用U-2.5% Nb合金试样在70 ℃下反应约5 min后,氢压就下降了3%,而对于纯铀[17],降低同样的氢压则需要更高的反应温度(125 ℃)和更长的反应时间(20 min)。由此可见,U-2.5% Nb合金的氢化速率远高于纯铀的,且一旦发生形核,则形核点迅速长大,停止反应后,即可得到氢化后试样。
由图6可见:类似于纯铀,氢化后U-2.5% Nb合金也具有点蚀特性,且在空间上分布不均匀;而不同于纯铀的是,U-2.5% Nb合金的氢蚀并不会形成弥散分布于试样表面的“小形核点”(不随反应时间的延长而长大),只形成“大形核点”(随着反应时间的延长而会长大),即U-2.5% Nb合金氢蚀后形成的形核点均迅速长大,呈圆点形,氢化点大小的差异代表了氢化初始形核时间的长短。进一步放大氢蚀点边缘区域可见:氢蚀点的边缘并非圆滑,而是呈条状向外凸出的。与图3相比,氢蚀点边缘外凸的条状宽度与氢蚀前U-2.5% Nb合金的贫铌α相宽度一致,均约为1 μm。可以推断在U-2.5% Nb合金发生氢蚀时,其表面的贫铌α相会优先腐蚀,而SEM形貌也证实了这一点。由图7(a)可见:由于氢化物的生成会造成体积膨胀,因而随着反应时间的延长,氢蚀点长大并破裂。进一步放大氢蚀点的边缘 (红色方框) 区域如图7(b),可以更直观地看出氢蚀点边缘呈外凸的条状,且外凸的条状正对应于U-2.5% Nb合金的α相,也就是说在U-2.5% Nb合金表面,贫铌α相的氢蚀速率要高于富铌γ1-2相的,优先发生氢化腐蚀,且随着反应时间的延长,相间于α相的γ1-2相也相继被腐蚀,从而使氢蚀点连成一片迅速长大。

(a) 低倍 (b) 高倍图6 U-2.5% Nb合金氢化后LSCM形貌Fig. 6 LSCM morphology of U-2.5% Nb alloy after hydriding at low (a) and high (b) magnification

(a) 低倍 (b) 高倍图7 U-2.5% Nb合金氢化后SEM形貌Fig. 7 SEM morphology of U-2.5% Nb alloy after hydriding at low (a) and high magnification
由图8可见:氢化前后试样均可以观察到贫铌α相和富铌γ1-2相的峰,且氢化后试样有明显的UH3峰。然而氢化生成的粉末UH3暴露于空气中很容易被氧化而不易被检测到。该试样之所以能检测到UH3峰,可能的原因有两个:一是该试样氢化时氢压下降了3%,氢化区域相对较大,氢蚀相对严重,生成的氢化物相对较多而仍有一部分未被氧化;二是U-2.5% Nb合金的氢蚀点比较深,试样内部生成的氢化物无法暴露于空气中发生氧化。
铀氢化的影响因素很多,且会在某些特殊位置优先形核,目前对此主要有两种不同观点:一种认为氧化膜的厚薄及完整性是决定铀氢化优先形核点的主导因素;另一种则认为试样本身尤其是其化学成分及微观组织结构是氢化优先形核的主导因素。
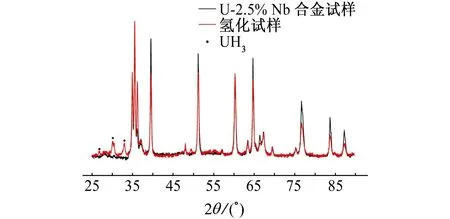
图8 氢化前后试样的XRD图谱Fig. 8 XRD patterns of the U-2.5% Nb alloy before and after hydriding
在本工作中,试样磨抛光亮后便立刻装入反应容器,尽量缩短了试样暴露于空气中的时间,氧化膜厚度已达几十个纳米,在后续对试样进行预热处理时,真空小于1 Pa时,160 ℃保温30 min,氧化膜进一步增厚,直至通氢,试样表面已覆盖一层较厚的氧化膜。通常认为在铀氢化反应初期,氢原子透过氧化膜“障碍”,抵达氧化膜与基体界面处,至氢浓度达到氢化反应的临界浓度时会发生氢蚀。然而对于U-2.5% Nb合金,由图3可以看出其中贫铌α相与富铌γ1-2相的耐蚀性不同,因而在试样表面形成的氧化膜并不平整,不同相之间氧化膜厚薄不均且在相界面处不连续。
图9为U-2.5% Nb合金氢化反应机理的可能示意图。有研究表明,α相的氧化速率远高于γ1-2相的,因而α相表面的氧化膜生长形成了表面浮凸,且向深度方向延伸,双相界面处氧化膜高低不平且不连续甚至存在物理通道。
在发生氢蚀时,相界面处表现为氢原子透过快速通道在氧化膜/α相界面处聚集,当达到氢化反应所需的临界浓度时优先在α相发生氢化形核[14]。随着形核点的长大,α相迅速腐蚀而生成大量氢化物,而产生的体积膨胀会引发氧化膜的变形。随着反应进一步加剧,相间于贫铌α相之间的γ1-2相也相继发生氢蚀而连成一片后迅速长大,相界面处氧化膜则发生了明显的扭曲甚至破裂,加速了氢的透过。这可能就是U-2.5% Nb合金氢蚀速率比纯铀高的原因之一。
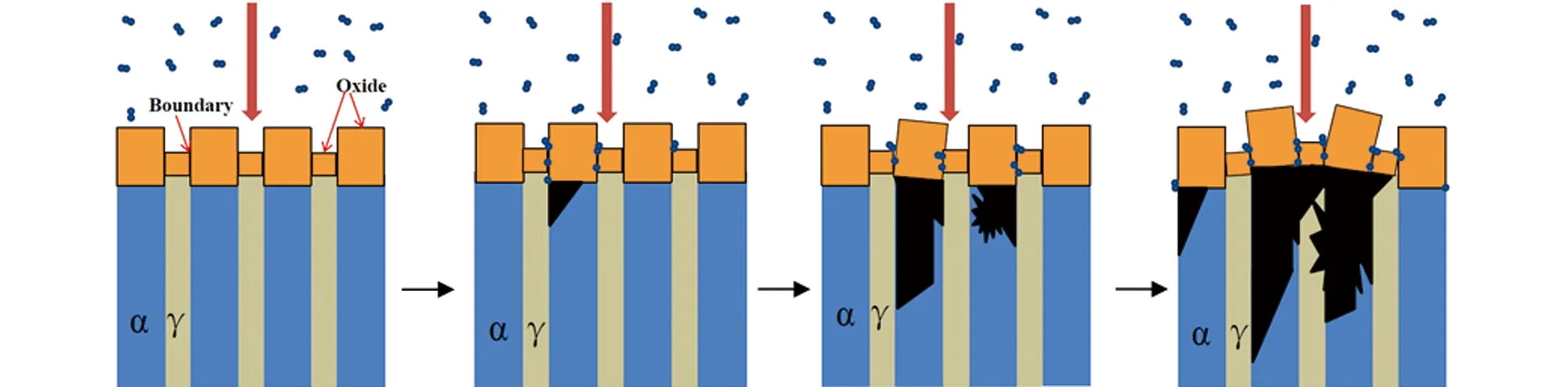
图9 U-2.5% Nb合金氢化反应的示意图Fig. 9 Scheme of the possible formation mechanism of hydriding reaction of U-2.5% Nb alloy
本试验结果表明,对于双相结构的U-2.5% Nb合金,氢化形核位点与组织结构之间有一定关联,U-2.5% Nb合金的氢蚀优先在贫铌α相发生,随后相间于α相的富铌γ1-2相相继被氢化,而后连成一片呈圆形迅速长大,这可能是由相结构及成分的不同造成的,后续工作会进一步探讨。
3 结论
(1) U-2.5% Nb合金的氢蚀很快,且只形成“大形核点”,即形核后均迅速长大。
(2) 在U-2.5% Nb合金表面,贫铌α相的氢蚀速率高于富铌γ1-2相的而优先发生氢化腐蚀,并随着反应时间的延长,相间于α相的γ1-2相也相继发生氢蚀而连成一片迅速长大。即U-2.5% Nb合金的氢蚀具有优先形核位点:贫铌α相。
参考文献:
[1] RITCHIE A G,GREENWOOD R C,RANDLES S J. The kinetics of the uranium-oxygen-water vapour reaction between 40 and 100 ℃[J]. Journal of Nuclear Materials,1986,139(2):121-136.
[2] MAGNANI M J. Reaction of uranium and its alloys with water vapor at low temperatures[M]. USA:Sandia Laboratory,1974,3.
[3] BALOOCH M,HAMZA A V. Hydrogen and water vapor adsorption on and reaction with uranium[J]. Journal of Nuclear Materials,1996,230(3):259-270.
[4] RITCHIE A G. The kinetic and mechanism of the uranium-water vapour reaction-an evaluation of some published work[J]. Journal of Nuclear Materials,1984,120(2):143-153.
[5] HASCHKE J M. Reactions of plutonium and uranium with water:kinetics and potential hazards[M]. Irvine:Los Alamos National Lab,1995.
[6] CALHOUN C A,GARLEA E,SISNEROS T,et al. Effects of hydrogen on the mechanical response of α-uranium[J]. Journal of Nuclear Materials,2015,465:737-745.
[7] BAZLEY S G,PETHERBRIDGE J R,GLASCOTT J,et al. The influence of hydrogen pressure and reaction temperature on the initiation of uranium hydride sites[J]. Solid State Ionics,2012,211:1-4.
[8] BLOCH J,SIMCA F,KROUP M,et al. The initial kinetics of uranium hydride formation studied by a hot-stage microscope technique[J]. Journal of Less Common Metals,1984,103(1):163-171.
[9] KNOWLES J P,FINDLAY I M,GEESON D A,et al. The influence of vacuum annealing on the nucleation and growth kinetics of uranium hydride[J]. MRS Online Proceedings,2012:1444(1):211-216.
[10] SCOTT T B,ALLEN G C,FINDLAY I,et al. UD3formation on uranium:evidence for grain boundary precipitation[J]. Philosophical Magazine,2007,87(2):177-187.
[11] ZHANG G,WANG X,WU J,et al. Influence of silicon impurity on the reaction of U-0.7% Ti alloy and hydrogen[J]. Journal of Alloys and Compounds,2015,648:122-126.
[12] DEMINT A L,LECKEY J H. Effect of silicon impurities and heat treatment on uranium hydriding rates[J]. Journal of Nuclear Materials,2000,281(2):208-212.
[13] STITT C A,PARASKEVOULAROS C,HARKEN N J,et al. The effects of metal surface geometry on the formation of uranium hydride[J]. Corrosion Science,2015,98:63-71.
[14] SHI P,SHEN L,BAI B,et al. Preferred hydride growth orientation of U-0.79% Ti alloy with β+U2Ti microstructure[J]. Journal of Nuclear Materials,2013,441(1/3):1-5.
[15] 纪和菲. 组织结构对U-5.7% Nb氢化反应的影响[D]. 绵阳:中国工程物理研究院,2013.
[16] LI R,WANG X. Effect of niobium additions on initial hydriding kinetics of uranium[J]. Journal of Nuclear Materials,2014,449(1):49-53.
[17] JI H,SHI P,LI R,et al. The microstructure and hydriding characteristics of high temperature aged U-13% Nb alloy[J]. Journal of Nuclear Materials,2015,464:43-47.
[18] KATZ O M,GULBRANSEN E A. Some observations on the uranium-niobium-hydrogen system[J]. Journal of Nuclear Materials,1962,5(3):269-279.
[19] OWEN L W. A microscope study of the initiation of hydrogen-uranium reaction[J]. Corrosion Science,1966,6:461-465.
[20] SHAMIR N,SCHWEKE D,RUBIN A,et al. Carbon enhanced hydrogen attack on an oxidized U-0.1% Cr surface[J]. Materials Science and Engineering,2010,9:012037.
