木聚糖酶基因的克隆与表达
2018-06-07宋立立顿宝庆张亚楠田景汉
宋立立, 顿宝庆, 张亚楠, 田景汉
(1.沧州师范学院生命科学学院,河北沧州 061000;2.中国农业科学研究院作物科学研究所/国家农作物基因资源与基因改良重大科学工程/生物质能源研究中心,北京 100081)
木聚糖是植物半纤维素的主要成份,是除纤维素之外自然界中最为丰富的多糖,也是自然界中最为丰富的可再生资源之一[1]。半纤维素在秸秆中仅次于纤维素,是可再生资源,但是大部分被作为废弃物浪费。我国秸秆资源丰富,将秸秆中的半纤维素转化为可利用资源是一个尚待解决的问题。木聚糖酶是木聚糖降解中最关键的酶,可以使木聚糖分子中β-1,4糖苷键断裂,水解产物为木二糖和木寡糖。木聚糖酶在能源工业、造纸制浆、食品、纺织、饲料等方面具有广阔的应用前景[2-3]。用木聚糖酶对含半纤维素的纸浆进行处理,可以减少漂白工艺中化学的用量,减轻对环境的污染,还可以提高纸浆的漂白度;木聚糖酶还可以作为饲料添加剂来提高饲料的利用率,改善饲料的营养价值;制酒工业添加木聚糖酶降低非淀粉多糖的含量,使后期易于过滤,酒色变澄清。国内已经成功构建了木聚糖酶工程菌株,但是和国外相比有着一定的差距,对木聚糖酶的研究较晚,因此,对木聚糖酶工程菌的进一步研究是有必要的。本研究通过分子生物学技术从桔青霉中克隆得到编码木聚糖酶基因,将其在毕赤酵母GS115和大肠杆菌BL21进行了表达研究,为木聚糖酶作为酶制剂应用提供理论依据。
1 材料与方法
1.1 材料
1.1.1 培养基 LB培养基参照《分子克隆实验指南》配制。酵母YPD培养基、选择MD培养基、诱导表达BMGY和BMMY培养基均根据Invitrogen公司毕赤酵母表达操作手册配制。
1.1.2 菌株与质粒 含有木聚糖酶的桔青霉菌株由笔者所在实验室筛选得到;克隆宿主菌EscherichiacoliDH5α、毕赤酵母菌GS115、大肠杆菌BL21为实验室保存。表达载体pPAL7购自BIO-RAD公司;pPIC9K购自Invitrogen公司。
1.1.3 主要酶和试剂 试验所涉及限制性核酸内切酶均购自NEB公司;PGEM-Teasy购自Promega公司;DNA回收、DNA纯化和DNA提取试剂盒均购自TaKaRa公司;DNA连接酶购买自Promega公司;质粒小量提取试剂盒购自天根公司;氨苄青霉素购自Sigma公司。其他试剂均为国产分析纯。
1.2 试验方法
1.2.1 缓冲液配制 配制方法参照《分子克隆实验指南》。
1.2.2 桔青霉CR-2的RNA提取 提取方法参照RNA提取试剂盒操作说明书。
1.2.3 RNA反转录cDNA 操作过程参照《分子克隆实验指南》。
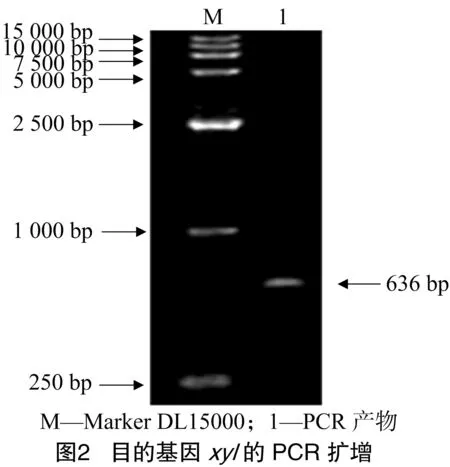
1.2.4 木聚糖酶基因片段的扩增 根据木聚糖酶基因cDNA序列设计引物,真核表达系统引物为P1:5′-TACGTAATGATTAAGTCTAA-3′;P2:5′-GAATTCTTACCA CACCGTTA-3′,下划线分别为SnaBⅠ和EcoRⅠ酶切位点序列。原核表达系统引物为P1:5′-CGTCCATATGATTAAG TCTAAAAAG-3′;P2:5′-GAATTCTTACCACACCGTTA-3′,下划线分别为NdeⅠ和EcoRⅠ酶切位点序列。扩增反应条件为94 ℃预变性5 min;94 ℃变性45 s,53 ℃退火45 s,72 ℃延伸2 min,30个循环;72 ℃延伸7 min。产物经1%琼脂糖凝胶电泳鉴定。
1.2.5 质粒提取 提取方法参照质粒小量提取试剂盒说明书。
1.2.6 转化大肠杆菌[4]
1.2.7 酶切验证 酶切反应体系(μL):Plasmid DNA 2 μL,10×enzyme Buffer 5 μL,SnaBⅠ 0.5 μL,EcoRⅠ 0.5 μL,补充dd H2O至20 μL。37 ℃过夜酶切(内切酶相应变化),酶切产物经琼脂糖凝胶电泳鉴定。
1.2.8 感受态制备 大肠杆菌DH5α和BL21感受态的制备参照文献[5];酵母菌株GS115感受态的制备参照文献[6]。
1.2.9 超声波破碎菌体 操作过程参照《分子克隆实验指南》。
1.2.10 酵母的电击转化 将9 μg的线性化好的DNA片段溶解在12 μL去离子水中,与80 μL的感受态混匀,转入电转化杯中4~10 ms进行电击;电击后,迅速加入1 mL 1 mol/L预冷的山梨醇溶液;电击产物放置30 ℃恢复30 min,涂于MD平板上,每450 μL涂布1块平板,将平板置于30 ℃培养,直至单个菌落出现。
1.2.11 表达质粒载体的线性化 转化前用ScaⅠ酶切,酶切体系(μL):pPIC9K-xyl80 μL,10×Buffer 20 μL,ScaI 10 μL,补加dd H2O至200 μL。
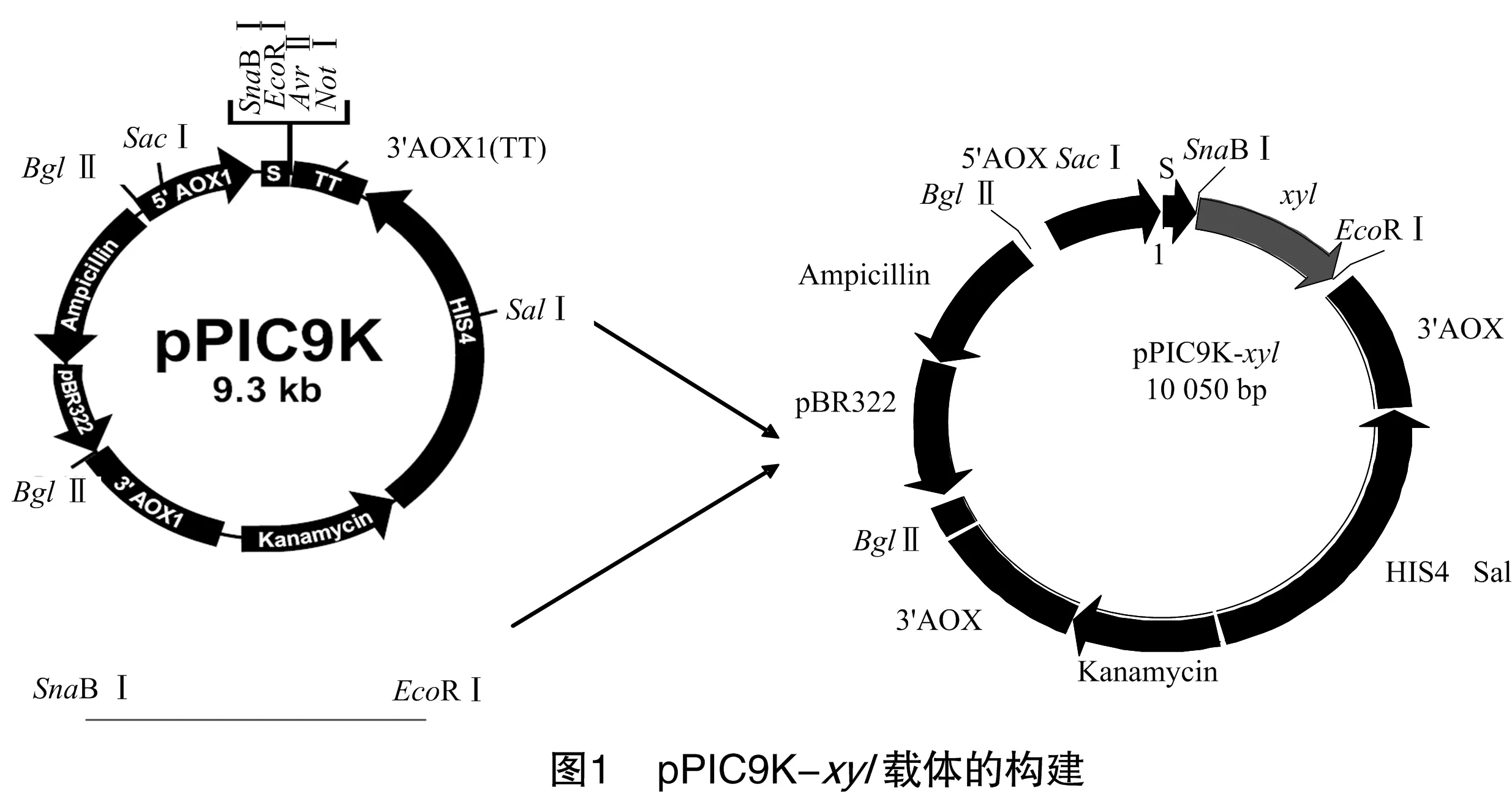
1.2.12 真核表达载体的构建 将连有xyl基因的克隆用LB液体培养基活化并提取质粒。用SnaBⅠ和EcoRⅠ双酶切质粒与pPIC9K载体,回收xyl片段及载体大片段,将2个回收产物连接构建表达载体pPIC9K-xyl,构建过程如图1。转化大肠杆菌DH5α,得到的转化子用SnaBⅠ和EcoRⅠ双酶切鉴定,鉴定正确的转化子用于下一步试验。
1.2.13 毕赤酵母的诱导表达试验 挑选阳性克隆于20 mL BMGY培养基摇瓶培养,28~30 ℃培养至D600 nm为2~6,离心5 min收集菌体,用1/5原培养体积的BMMY重悬菌体;将菌液重新置于100 mL的摇瓶28~30 ℃摇床培养;每隔 24 h 向培养基中添加无水甲醇至终浓度为1.0%;隔24 h取样,128 000 r/min 离心5 min,收集上清,分析重组菌的活性。取样时间一般为0、24、48、72、96、120 h。
1.2.14 大肠杆菌的诱导表达试验 挑选正确的菌落,接入10 mL含100 mg/mL氨苄青霉素LB培养液中,37 ℃培养至D600 nm为0.2~0.6;取100 μL 样品加入50 mL含100 mg/mL氨苄青霉素LB培养液中,37 ℃培养至D600 nm为0.2~0.6,加IPTG至终浓度0.8 mmol/L,37 ℃培养,每2 h取样,样品超声波破碎后分析其胞内表达情况,利用SDS-PAGE等活性试验检测与鉴定重组蛋白的表达。
1.2.15 重组蛋白SDS-PAGE检测 重组蛋白SDS-PAGE检测操作步骤见参考文献[7],蛋白电泳主要试剂配制方法见参考文献[8-9]。
1.2.16 木聚糖酶活性测定方法 配制10.0 mg/mL的木糖溶液:称取无水木糖1.000 g,加入去离子水后定容至 100 mL。取8支试管按表1加入试剂,稀释木糖标准液,再加入3 mL DNS试剂;充分摇匀置于沸水中煮5 min;流动冷水冷却后,将显色液以4 000 r/min离心5 min。取上清液,用0号试管作对照,在540 nm下测其他试液的吸光度D540 nm。以吸光度为纵坐标,以木糖浓度为横坐标绘制标准曲线。
木聚糖酶活性测定方法:取3支试管各加0.5 mL中性木聚糖底物,与待测酶液一起在50 ℃水浴中预热5 min。在第1、2试管中加入0.5 mL待测酶液,50 ℃水浴中反应15 min。加入3 mL的DNS试剂,在第3支试管中加入0.5 mL的待测酶液,沸水浴5 min。将显色液(包括空白)以4 000 r/min离心5 min。取上清夜以第3支试管为对照在540 nm条件下测第1、 2试管样的吸光度D540 nm。吸光度以在0.2~0.3为宜,若不在此范围可改变稀释倍数重做。

表1 酶活性测量用木糖标准曲线溶液配制
酶活性的计算:酶活性(IU/mL或IU/g)=木聚糖等量值÷150÷15÷0.5×n。
式中:150指木糖从微克量换算成微克分子数;15指待测液与底物的反应时间;0.5指与底物反应的待测酶液量;n指原酶液(或固体原酶)的稀释倍数
2 结果与分析
2.1 目的基因的片段克隆结果
PCR产物电泳鉴定结果表明,在636 bp的位置有一明显条带,与公布的基因片段大小相一致(图2)。回收扩增片段连接pGEM-Teasy测序,所测序列与GenBank中报道的木聚糖酶基因序列同源性为99.99%,编码212个氨基酸。将所得到的木聚糖酶基因作原核表达和真核系统表达,重组质粒pGEM-xyl经SnaBⅠ和EcoRⅠ(真核表达)、NdeⅠ和EcoRⅠ(原核表达)双酶切验证,酶切后电泳鉴定出2条带:一条带为636 bp,与PCR产物带相同;另一条带为3 000 bp,与载体pEGM-Teasy的条带大小一致(2个图大小相一致,以真核表达系统酶切图为例,图3)。结果表明,初步获得了木聚糖酶编码基因xyl。

2.2 表达载体的构建
原核表达载体(pPAL7)和真核表达载体(pPIC9K)的构建方法一致,同时双酶切载体质粒和pGEM-xyl阳性转化子质粒,酶切产物同时回收,回收产物和表达载体过夜酶连,连接后转化大肠杆菌DH5α;重组子经双酶切验证,在真核表达载体中,质粒经SnaBⅠ和EcoRⅠ双酶切电泳检测如图4所示,在636、9 000 bp有2条明显的条带,分别与扩增产物和载体大小相一致;在原核表达系统中,重组子质粒经NdeⅠ和EcoRⅠ双酶切电泳检测,在636、5 641 bp处有2条与预期大小相一致的条带(图5),初步表明,该重组子含有木聚糖酶编码基因。验证正确的质粒分别转化大肠杆菌BL21和酵母GS115进行诱导表达试验。


2.3 木糖酶在原核系统和真核系统中的表达分析
2.3.1 SDS-PAGE分析(原核表达系统) 用IPTG对重组菌进行不同时间的诱导,表达产物进行SDS-PAGE电泳分析,在22 ku处呈现表达蛋白条带,与理论蛋白大小相一致(图6),根据诱导时间与表达量的关系,37 ℃条件下诱导6 h蛋白表达量最高。

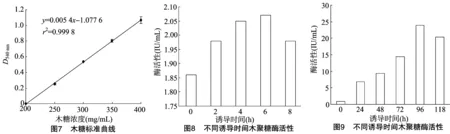
2.3.2 木糖标准曲线 由图7可知,标准曲线方程为y=0.005 4x-1.077 6(x轴为木糖浓度,y轴为D540 nm),根据标准方程可以计算出还原糖浓度,根据木聚糖酶酶活性计算公式求出酶活性,酶活性单位为IU/mL。
2.3.3 木聚糖酶基因在原核系统中的表达 在原核表达系统中,重组子经IPTG诱导,每隔2 h取样,测定其酶活性(图8),诱导6 h时酶活性最高,达到2.07 IU/mL。
2.3.4 木聚糖酶基因在真核系统中的表达 在真核表达系统中,重组子经甲醇诱导,每间隔24 h取样,测定其酶活性,诱导96 h时酶活性最高,达到23.84 IU/mL(图9)。
2.3.5 木聚糖酶基因氨基酸保守序列分析 根据NCBI比对木聚糖酶氨基酸的保守序列分析,该酶属于11家族糖基水解酶,保守序列有28~211个氨基酸序列,与许多菌种的氨基酸保守序列同源性很高(图10)。
3 小结
根据已经报道的桔青霉菌中木聚糖酶基因的同源序列设计引物,提取桔青霉的RNA,通过RT-PCR扩增得到木聚糖酶的编码基因xyl,结果进行比对分析,阅读框编码212个氨基酸,表达蛋白分子量大小为22 ku。
木聚糖酶的表达活性分析中,同时测定了在原核系统和真核系统中酶的表达量,发现木聚糖酶在真核表达系统中的酶活性(23.84 IU/mL)远远要高于原核表达系统中的酶活性(2.07 IU/mL),推测可能原因为该基因来源于真核生物,所以在真核系统中更容易实现表达。本试验研究木聚糖酶基因的克隆与表达,该工程菌在没有对载体和基因改造的情况下,对产酶条件未进行优化,表达量不是很高。本研究侧重克隆表达,后续的研究中可通过对该基因和载体进行改造、优化其产酶条件来提高酶的活性。

参考文献:
[1]杨梦华,李 颖,关国华,等. 极端耐热木聚糖酶基因在大肠杆菌和毕赤酵母中的高效表达[J]. 微生物学报,2005,45(2):236-239.
[2]张桂敏,饶 犇,叶 戋,等. 棉花黄萎病真菌Verticilliumdahliae木聚糖酶基因的克隆、表达和酶学性质分析[J]. 微生物学报,2008(6):765-771.
[3]Biely P,Vrsanska M,Tenkanen M,et al. Endo-β-1,4-xylanasfamilies:differences in catalytic properties[J]. Journal of Biotechnology,1997,57(1/2/3):151-166.
[4]杨立泉,马春晓,戴晓军,等. 一个新的葡萄糖淀粉酶基因在大肠杆菌中的分泌表达[J]. 生物学通报,2007,42(3):55-57.
[5]许正宏. 微生物耐碱性木聚糖酶的合成、调控及底物降解方式的研究[J]. 无锡:江南大学,2005.
[6]张八生. 东亚飞蝗和金龟子绿僵几丁质基因的克隆与表达研究[D]. 重庆:重庆大学,2008.
[7]曲小爽. 高效桔青霉纤维素分解菌筛选及酶学初探[D]. 哈尔滨:东北农业大学,2008.
[8]郭尧君. 蛋白质电泳实验技术[M]. 北京:科学出版社,2005:54-157.
[9]赵永芳. 生物化学技术原理及其利用[M]. 北京:科学出版社,2000:1-502.
