氯化钠吸热条件下的纳米硅制备及其电化学性能
2018-05-22
(湖南大学 材料科学与工程学院,湖南 长沙 410082)
随着电子设备和电动汽车行业的快速发展,人们对可充电电池的需求日益增加。追求容量高、倍率性能优异及循环性能稳定的可充电电池是当前研究的热点[1-2]。硅因具有最高的理论比容量 3580 mAh·g–1(Li3.75Si,室温下形成),稳定的充电平台(约0.45 V vs. Li/Li+)、安全无毒并且储量丰富等优势,成为最有应用前景的下一代锂离子电池负极材料之一。然而,由于硅电极在充放电过程中伴随着较大的体积效应,导致硅颗粒发生粉化、破裂以及活性材料的脱落。另外,硅电极在循环过程中不断消耗电解质,形成固体电解质界面(SEI)膜[3-6],致使其容量快速衰减,循环稳定性能变差,从而限制了硅材料的商业化应用[7-8]。为了解决体积效应问题,研究者们做出大量的尝试,包括制备超细纳米硅(<50 nm),开发新型粘结剂、设计新型纳米硅材料,例如硅纳米颗粒[9]、硅纳米线[10]、硅纳米管[11]、硅空心纳米球[12]、核-壳结构[13]等,有效缩短锂离子的扩散路径,缓解体积效应,改善循环稳定性。Zhou等[14]利用天然高岭土作为原料,通过选择性酸腐蚀和镁热还原的方法成功制备出纳米硅材料。该材料由直径为20~50 nm的晶粒相互连接而成,这种纳米晶粒组成的多孔结构使得该材料具有非常优良的电化学性能,在0.2C倍率下循环100次,可以获得高达2200 mAh·g–1的稳定比容量,但是该材料首次充放电库伦效率较低,仅为79.2%。Liu等[15]利用镁热还原稻壳制备出具有多孔结构的纳米硅,该多孔材料具有纳米尺度的孔隙和孔壁,并且稻壳资源丰富、廉价易得。但是在30 mA·g–1电流密度下循环50次后,可逆比容量保持率仅为23.3%。
众所周知,镁热还原法是一种低成本、可扩展的制备硅材料的方法。自2007年以来,通过镁热还原法还原不同的硅源已制备出多种硅纳米结构,其中有纳米晶体、纳米管、纳米线等[16-18]。然而,由于镁热还原过程是一个自发放热反应,使得实验过程中局部温度达到 1941 ℃[19],远远超过了加热温度,导致硅晶粒过度生长,难以得到纳米硅材料。本文利用氯化钠(熔点:801 ℃)作为吸热剂,分别以气相二氧化硅和金属镁作为硅源和还原剂,通过镁热还原法成功制备出纳米硅材料,该方法获得较高的收率,达到 82%。在不经过任何修饰的情况下,对纳米硅进行电化学性能测试,首次以 0.1 A·g–1的电流密度活化,测得脱锂比容量高达 1875 mAh g–1。以0.6 A·g–1电流密度循环50次后,脱锂比容量仍保持 1007 mAh·g–1。
1 实验
1.1 纳米硅的制备
首先将气相二氧化硅和氯化钠[质量比(SiO2/NaCl)=1:1]混合均匀,再与适量镁粉进行混合,并转移到刚玉坩埚里,放置于管式炉中,在高纯氩气的环境下以10 ℃/min的升温速度加热至650 ℃,保温2 h,冷却至室温后取出产物。其次将产物先用去离子水清洗,通过干燥滤液回收氯化钠,接着分别用质量分数为5%HCl和5%HF清洗,除去反应产物MgO、过量的SiO2和少量的副产物Mg2SiO4。最后采用去离子水清洗过滤,然后将样品放入真空干燥箱60 ℃下干燥过夜,即得到纳米硅。为了对比氯化钠吸热剂对纳米硅电化学性能的影响,在相同的实验条件下未添加吸热剂制备纳米硅,分别命名为SC-Si和 Si。
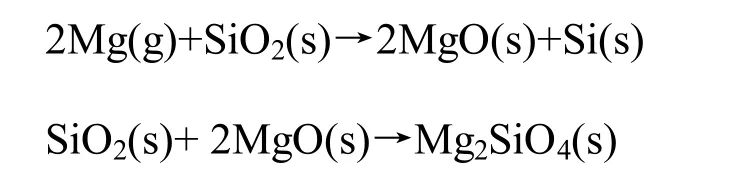
1.2 样品表征
通过X射线衍射仪(Philips X’Pert SUPER)对制备的纳米硅进行物相分析。用扫描电镜(JEOL.JSM.6700F)和透射电镜(Hitachi H7650:JEOL 2010)对纳米硅进行形貌结构表征。
1.3 电化学性能测试
将制备的纳米硅作为活性材料制成工作电极。纳米硅、导电炭黑和粘结剂海藻酸钠按质量比6:2:2混合均匀,溶剂采用去离子水,磁力搅拌制成均匀浆料,涂覆在预先用无水乙醇洗过的铜箔上。随后在80 ℃真空条件下干燥10 h,自然冷却至室温后,用切片机切成φ=14 mm的电极片。
在填充高纯氩气的手套箱内(H2O,O2体积分数<l0–6)进行电池组装,以高纯锂片作为对电极,聚丙烯微孔薄膜(Celgard 2400)作为隔膜,以1 mol·L–1的六氟磷酸锂(LiPF6)/碳酸乙烯酯(EC)/碳酸二乙酯(DEC)/添加2%(体积分数)碳酸亚乙烯酯(VC)作为电解液(珠海赛维电子材料有限公司),组装成CR2016型纽扣电池。制备得到的纽扣电池放置于室温干燥环境中静置若干小时后进行测试。采用新威尔(深圳)高精度电池性能测试系统(5 V-1 mA)对电池比容量进行测试,充放电电压为 0.05~1.0 V,电流密度为0.6 A·g–1。循环伏安测试和电化学阻抗测试在电化学工作站 (CHI604E,上海辰华)上进行,循环伏安参数:电压范围为0.01~1.0 V,0.1 mV·s–1;电化学阻抗参数:频率范围为10–2~105Hz,测试振幅为5 mV。
2 结果与讨论
2.1 纳米硅的XRD分析
图1分别为添加氯化钠吸热剂和未添加吸热剂制备的纳米硅XRD谱,均在2θ为28.4°,47.3°,56.1°,69.3°位置出现纳米硅的衍射峰,分别对应于(111)、(220)、(311)、(400)晶面,并且与 JCPDS卡片No.27-1402相对应。由图可知,与添加氯化钠吸热剂制备纳米硅的衍射峰相比,未添加吸热剂制备纳米硅的衍射峰更加尖锐,表明该条件下制备晶体硅的结晶度更高,晶粒的粒径更大。通过谢乐公式计算(111)晶面对应的数据可得,加氯化钠吸热剂制备的纳米硅晶粒平均粒径为25 nm,未添加吸热剂制备的纳米硅晶粒平均粒径为64 nm。说明在实验过程中添加氯化钠能够吸收掉镁热还原过程放出的大量热量,从而制备出粒径较小的纳米硅。在不考虑任何热损耗的情况下,经热力学计算,添加氯化钠吸热剂使得镁热还原反应局部温度低于1310 ℃,而未加氯化钠吸热剂的镁热还原反应局部温度达到1941 ℃,因此采用氯化钠吸热剂有利于制备出粒径较小的纳米硅材料。
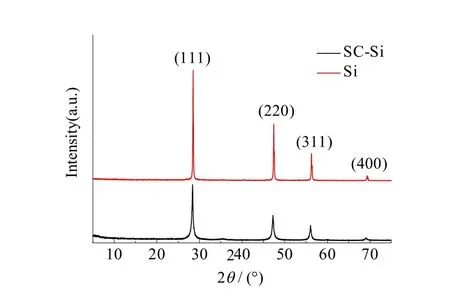
图1 SC-Si和Si的XRD谱Fig.1 XRD patterns of SC-Si and Si samples
2.2 纳米硅的TEM和HRTEM分析
添加氯化钠吸热剂制备纳米硅的结构和形貌表征如图2所示,图2(a)为纳米硅的扫描电镜图,显示出纳米硅具有不规则的形貌和非常粗糙的表面。图2(b)为纳米硅的透射电镜图,由图可知,纳米硅是由许多平均粒径为25 nm的晶粒组成的类球形颗粒。图2(c)为纳米硅的选区电子衍射图,显示出硅的衍射环与XRD结果完全对应。另外,高分辨透射电镜图提供了更加精细的纳米硅结构特点,从图2(d)的晶格条纹中观察到大约0.32 nm的晶面间距,这与晶体硅的(111)晶面相对应。

图2 SC-Si的(a)扫描电镜图;(b)透射电镜图;(c)选区电子衍射图和(d)高分辨率透射电镜图Fig.2 SEM image (a); TEM image (b); electron diffraction pattern of a selected area (c) and HRTEM image of the SC-Si (d)
2.3 纳米硅的杂质元素测定
由于氯离子对电池正、负极材料具有腐蚀作用,造成电池比容量下降,严重的情况会造成极片穿孔,破坏整个电池。因此采用原子吸收光谱(AAS)对氯化钠吸热条件下制备的纳米硅进行测试,检测结果如表1,最终获得的纳米硅材料中残留钠离子质量分数0.024%,氯离子质量分数 0.026%。参照锂离子电极材料有关标准[20-21],本文所制纳米硅中钠离子和氯离子的含量在锂离子电极材料杂质含量允许范围之内。

表1 SC-Si中的杂质元素含量Tab.1 The content of impurity in SC-Si
2.4 纳米硅的电化学性能分析
图3(a)为添加氯化钠吸热剂制备纳米硅的充放电曲线图。具体测试条件为工作电压0.05~1.0 V(vs Li/Li+),首次循环以0.1 A·g–1的电流密度进行活化,第2次及以后以0.6 A·g–1电流密度进行循环测试。在材料的首次放电过程中,均在0.1 V附近有一个较长的电压平台,对应于晶体Si相转变为无定型LixSi相的过程。而充电过程中均在0.45 V处出现稳定的电压平台,对应于无定型LixSi相向无定型Si相转变的过程。进一步分析发现第一次放电曲线的电压平台与后续的放电曲线电压平台明显不一样,这主要是因为经过首次充放电循环后,晶体硅转变成无定型硅,随后的循环过程是无定型硅相的嵌脱锂过程[22-23]。由图可知,添加氯化钠吸热剂制备的纳米硅首次嵌、脱锂比容量分别为2215 mAh·g–1,1875 mAh·g–1,即首次库伦效率为85%,造成首次循环过程中不可逆容量的损失,主要是由于电解质在0.1 V附近发生分解,导致硅颗粒表面SEI膜的形成,消耗了部分锂离子。随着循环次数的增加,由于体积变化引起的应力-应变,致使纳米硅的容量持续下降,但速率较慢,这主要归结于氯化钠吸收掉镁热还原过程中放出的热量,抑制硅晶粒的过度生长,得到粒径较小的晶粒,从而缩短锂离子的传输路径,提高了纳米硅的利用率。
图3(b)为纳米硅的循环性能图。从图可知,添加氯化钠吸热剂制备纳米硅的首次脱锂比容量可达1875 mAh·g–1,接着以 0.6 A·g–1电流密度循环 50次后,脱锂比容量仍可保持1007 mAh·g–1,保持率达到 54%。而且在循环过程中库伦效率保持在98%~100%范围内,展现出良好的循环稳定性和可逆性。
图3(c)为添加氯化钠吸热剂制备纳米硅的倍率性能图,测试电流密度从0.1 A·g–1逐步上升到1.2 A·g–1再回到 0.1 A·g–1。在电流密度为 0.1 A·g–1时,测得纳米硅脱锂比容量达到1983 mAh·g–1,随着电流密度逐步上升到0.4,0.8,1.2 A·g–1时,对应的脱锂比容量依次降为1650,1017,675 mAh·g–1。然而,当电流密度恢复到0.1 A·g–1时,脱锂比容量能够恢复至1492 mAh·g–1。此结果表明添加氯化钠吸热剂制备的纳米硅具有优异的倍率性能,该电极材料经过高电流密度循环后仍可恢复至低电流密度的比容量和循环性能。
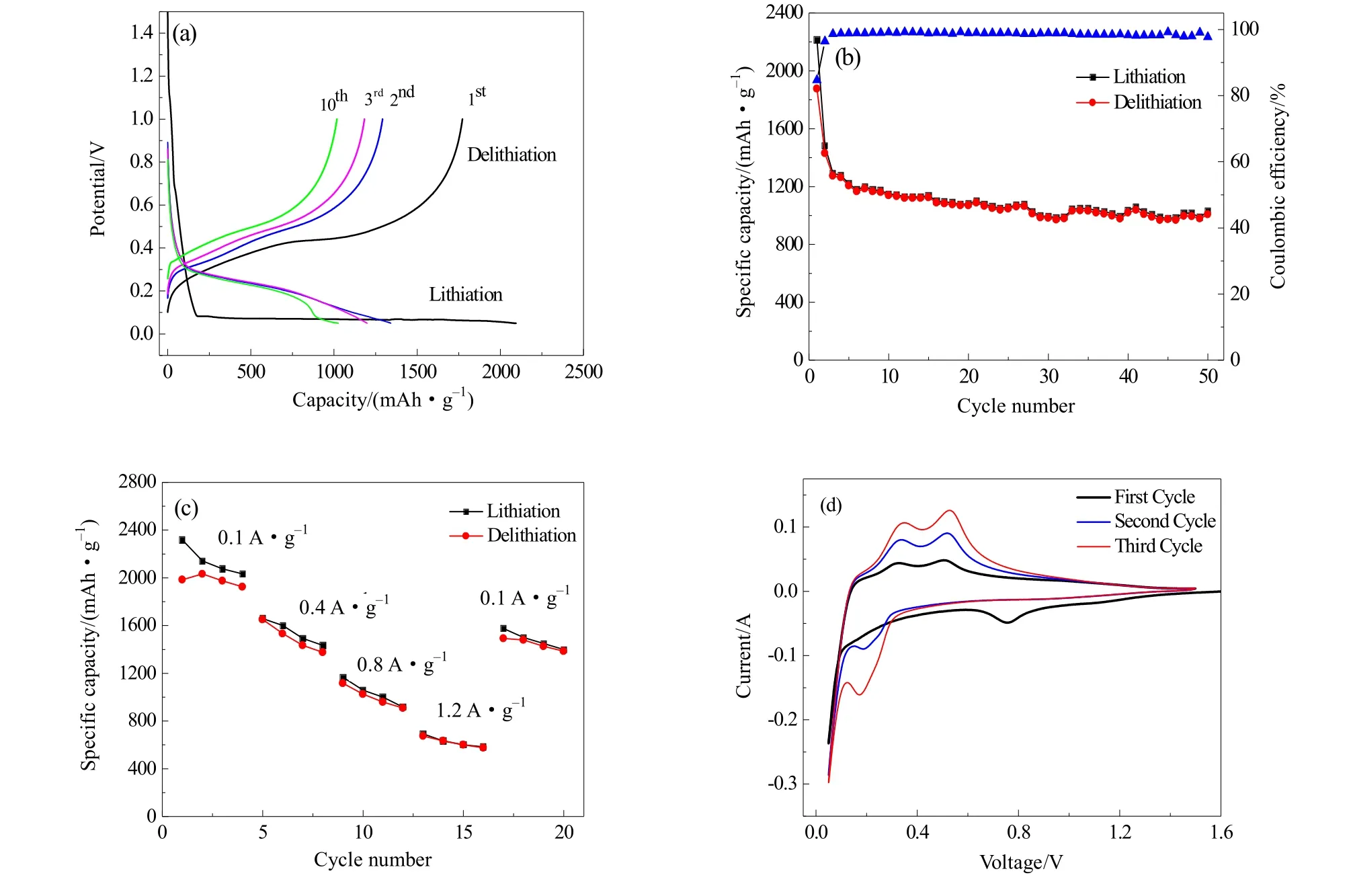
图3 SC-Si电极的(a)充放电曲线;(b)循环性能和库伦效率曲线;(c)倍率性能曲线;(d)循环伏安曲线Fig.3 The charge/discharge profiles at typical cycles of SC-Si electrode (a); cycling performance and columbic efficiency of SC-Si electrode (b); rate performance curves (c) and CV curves (d) of SC-Si electrode
图3(d)为添加氯化钠吸热剂制备的纳米硅1,2,3次循环伏安曲线,以0.1 mV·s–1扫描速率测试。由图可见,首次阴极扫描在0.78 V左右出现一个还原峰,这对应于电解液的分解和硅颗粒表面SEI膜的生成[24],而且在随后的扫描过程中消失,说明生成稳定的SEI膜,从而阻止电解质的进一步分解。电压降到0.2 V左右时阴极电流迅速增大,并且在接近0 V时达到峰值,这对应于纳米硅的首次合金化反应,晶体硅逐渐转变为LixSi无定型态的过程。首次阳极扫描过程中,分别在0.33 V和0.52 V的附近出现两个氧化峰,这对应纳米硅的首次去合金化反应,由无定型LixSi相转变为非晶态Si相的过程[25]。在第二次合金化过程中,在0.2 V附近出现较强的还原峰,对应非晶态Si相转变为LixSi合金相的过程,去合金化过程与首次相似,但是氧化峰值更高,说明经过首次循环后活性材料得到进一步活化,并且电极中锂离子扩散速率得以提高。
图4为纳米硅的交流阻抗图,对比添加氯化钠吸热剂和未添加吸热剂制备纳米硅的交流阻抗性能。由图可知,两种条件下制备的硅电极电化学交流阻抗谱图均是由一个高频区半圆弧和一段低频区斜线组成。其中高频区半圆弧对应于电解液与电极表面的界面阻抗,即SEI膜阻抗;低频区的斜线对应于在电极中离子的扩散阻抗。对比发现,添加氯化钠吸热剂制备的硅电极在高频区半圆弧的半径明显小于未添加吸热剂制备的硅电极,说明添加氯化钠吸热剂制备的纳米硅具有丰富的离子通道和电荷转移通道,因而电池的SEI膜阻抗和电荷转移阻抗得以减小。
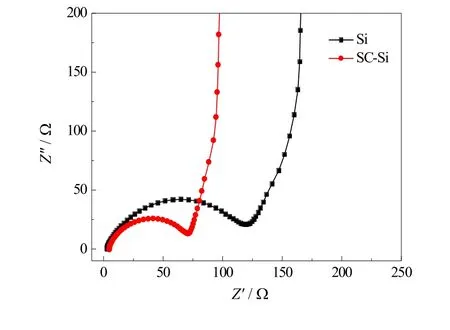
图4 SC-Si和Si电极循环前的交流阻抗图Fig.4 The impedance diagram of SC-Si and Si electrodes
3 结论
以氯化钠作为吸热剂,通过镁热还原气相二氧化硅成功制备出纳米硅材料。该材料在 0.1 A·g–1电流密度下测试,首次脱锂比容量达到 1875 mAh·g–1,首次库伦效率可达 85%,以 0.6 A·g–1电流密度循环 50次后,脱锂比容量仍可保持 1007 mAh·g–1。该纳米硅材料优良的电化学性能主要归因于在制备过程中氯化钠吸收掉镁热还原过程放出的大量热量,抑制了硅晶粒的过度生长,得到粒径较小的晶粒,有效地缩短锂离子的传输路径,从而使纳米硅的利用率得以提高。另外,本文所采用的方法制备纳米硅收率达82%,工艺简单、成本低廉,适合规模化生产。
参考文献:
[1]CHOI N S, CHEN Z, FREUNBERGER S A, et al.Challenges facing lithium batteries and electrical double-layer capacitors [J]. Angew Chem, 2012, 51(40):9994.
[2]XU J, WANG X F, WANG X W, et al. Three-dimensional structural engineering for energy-storage devices: from microscope to macroscope [J]. ChemElectroChem, 2014,1(6): 975-1002.
[3]KASAVAJJULA U, WANG C, APPLEBY A J. Nano- and bulk-silicon-based insertion anodes for lithium-ion secondary cells [J]. J Power Sources, 2007, 163(2):1003-1039.
[4]LIU X H, ZHONG L, HUANG S, et al. Size-dependent fracture of silicon nanoparticles during lithiation [J]. ACS Nano, 2012, 6(2): 1522-1531.
[5]MCDOWELL M T, RYU I, LEE S W, et al. Studying the kinetics of crystalline silicon nanoparticle lithiation with in situ transmission electron microscopy [J]. Adv Mater, 2012,24(45): 6034.
[6]GU M, LI Y, LI X, et al. In situ TEM study of lithiation behavior of silicon nanoparticles attached to and embedded in a carbon matrix [J]. ACS Nano, 2012, 6(9): 8439-8447.
[7]LIU H K, GUO Z P, WANG J Z, et al. Si-based anode materials for lithium rechargeable batteries [J]. J Mater Chem, 2010, 20(45): 10055-10057.
[8]HUGGINS R A. Lithium alloy negative electrodes [J]. J Power Sources, 1999(1/2): 13-19.
[9]LI H. A high capacity nano-si composite anode material for lithium rechargeable batteries [J]. Cheminform, 1999, 2(11):547-549.
[10]CUI L F, RUFFO R, CHAN C K, et al.Crystalline-amorphous core-shell silicon nanowires for high capacity and high current battery electrodes [J]. Nano Lett, 2009, 9(1): 491-495.
[11]ZHOU Y, JIANG X, CHEN L, et al. Novel mesoporous silicon nanorod as an anode material for lithium ion batteries [J]. Electrochim Acta, 2014, 127(5): 252-258.
[12]YAO Y, MCDOWELL M T, RYU I, et al. Interconnected silicon hollow nanospheres for lithium-ion battery anodes with long cycle life [J]. Nano Lett, 2011, 11(7): 2949-2954.
[13]LIU N, WU H, MCDOWELL M T, et al. A yolk-shell design for stabilized and scalable Li-ion battery alloy anodes [J]. Nano Lett, 2012, 12(6): 3315.
[14]ZHOU X, WU L, YANG J, et al. Synthesis of nano-sized silicon from natural halloysite clay and its high performance as anode for lithium-ion batteries [J]. J Power Sources, 2016, 324: 33-40.
[15]刘树和, 赵淑春, 姚耀春, 等. 稻壳制备锂离子电池硅负极材料 [J]. 昆明理工大学学报(自然科学版), 2014(6):14-18.
[16]KIM K H, DONG J L, CHO K M, et al. Complete magnesiothermic reduction reaction of vertically aligned mesoporous silica channels to form pure silicon nanoparticles [J]. Sci Rep, 2015, 5: 9014.
[17]JIA H P, GAO P F, YANG J, et al. Novel three-dimensional mesoporous silicon for high power lithium-ion battery anode material [J]. Adv Energy Mater, 2011(6): 1036-1039.
[18]FAVORS Z, BAY H H, MUTLU Z, et al. Towards scalable binderless electrodes: carbon coated silicon nanofiber paper via Mg reduction of electrospun SiO2nanofibers [J]. Sci Rep, 2015, 5: 8246.
[19]WON C W, NERSISYAN H H, WON H I. Solar-grade silicon powder prepared by combining combustion synthesis with hydrometallurgy [J]. Solar Energy Mater Solar Cells, 2011, 95(2): 745-750.
[20]中华人民共和国国家质量监督检验检疫总局. 中国国家标准化管理委员会. 钴酸锂: GB/T 20252-2006 [S]. 北京:中国标准出版社, 2006.
[21]中华人民共和国国家质量监督检验检疫总局. 中国国家标准化管理委员会. 锂离子电池石墨类负极材料: GB/T 24533-2009 [S]. 北京: 中国标准出版社, 2010.
[22]LI J, DAHN J R. An in situ X-ray diffraction study of the reaction of Li with crystalline Si [J]. J Electrochem Soc,2007, 154(3): A156-A161.
[23]OBROVAC M N, KRAUSE L J. Reversible cycling of crystalline silicon powder [J]. J Electrochem Soc, 2007,154(2): A103-A108.
[24]LIU W R, GUO Z Z, YOUNG W S, et al. Effect of electrode structure on performance of Si anode in Li-ion batteries: Si particle size and conductive additive [J]. J Power Sources, 2005, 140(1): 139-144.
[25]ZHANG L, HAO W, WANG H, et al. Porous graphene frame supported silicon@graphitic carbon via in situ solid-state synthesis for high-performance lithium-ion anodes [J]. J Mater Chem A, 2013(26): 7601-7611.
