Challenges and trend s in ap om orp hine d rug d elivery system s for the treatm ent of Parkinson’s disease
2018-05-15NrupBorkrHuilingMuRenHolm
Nrup Borkr,Huiling Mu,RenéHolm
a Department of Pharmacy,Faculty of Health and Medical Sciences,University of Copenhagen,Universitetsparken 2,Copenhagen,2100,Denmark
b Drug Product Development,Janssen Research and Development,Johnson&Johnson,Turnhoutsew eg 30,Beerse,2340,Belgium
Keywords:Apom orphine Drug delivery Parkinson’s disease Alternative apom orphine therapy Non-invasive delivery Excipients
A B S T R A C TParkinson’s disease(PD)is a chronic debilitating disease affecting approxim ately 1%of the population over the age of 60.The severity of PD is correlated to the degree of dopam inergic neuronal loss.Apom orphine has a sim ilar chem ical structure as the neurotransm itter dopam ine and has been used for the treatm ent of advanced PD patients.In PD patients,apom orphine is norm ally adm inistered subcutaneously w ith frequent injections because of the compound’s extensive hepatic f irst-pass metabolism.There is,hence,a large unmet need for alternative adm inistrative routes for apom orphine to im prove patient com pliance.The present review focuses on the research and developm ent of alternative delivery of apom orphine,aim ing to highlight the potential of non-invasive apom orphine therapy in PD,such as sublingual delivery and transdermal delivery.
1. Introd uction
The earliest reported synthesis of apomorphine was described by Arppe in 1845 and later by Matthiesen and Wright in 1869,w hich involved reacting m orphine w ith hydrochloric acid or sulfuric acid,respectively,to obtain apom orphine[1].Apom orphine found its early use in veterinary therapeutics to treat issues associated w ith farmyard anim al behavior.By 1874,it w as known that apomorphine had effects on the central nervous system along w ith em etic effects[1,2].In hum an m edical use,the com pound w as recom m ended as an em etic,sedative,as w ell as a treatm ent for narcotic and alcohol addiction[3].Weil in 1884 suggested apom orphine for its potential use in treating Parkinson’s disease(PD)[4,5].Schwab and cow orkers and Cotzias conf irm ed Weil’s suggestion,as they found a signif icant decrease in trem ors and rigidity associated w ith PD in hum ans[6–9].In today’s m edical treatm ent,subcutaneous apomorphine is used for the treatm ent of advanced PDin patients undergoing m otor disabilities that do not respond to other PD treatm ents.There is a large unm et need for alternative apom orphine delivery system s,as this treatm ent is provided by multiple subcutaneous injections daily.This review covers the im portance of apom orphine in PD m anagem ent and emphasizes the challenges w ith apom orphine therapy for PD patients.The present review also aim s at highlighting som e key research developments in the area of non-invasive delivery of apom orphine.
2. Physicochem ical properties of apom orphine
Apomorphine(6-methyl-6αβ-noraporphine-10,11-diol)belongs to the class ofβ-phenylethylam ines sharing structural sim ilarities to the neurotransm itter,dopam ine,both having a catechol m oiety(Fig.1).Apom orphine is a chiral m olecule(MW:267.32 Da).The R-form is a dopamine agonist,w hile the S-form of the m olecule m ay possess anti-dopam inergic activity[10,11].
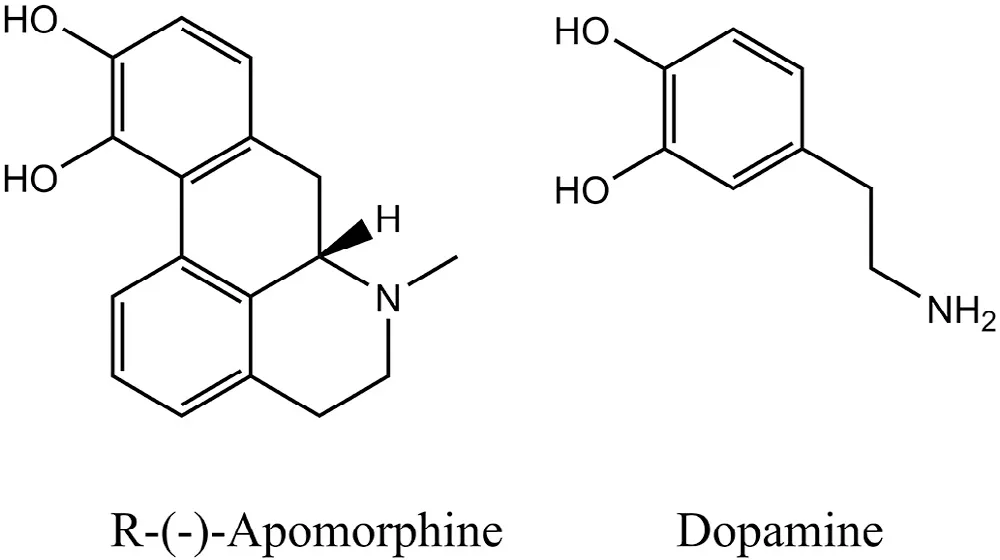
Fig.1–Chem ical structures of R-(-)-apom orphine and dop am ine.
Apom orphine is w ater-soluble w ith an interm ediate lipophilicity represented by a log P of 2.0[12].The com pound has tw o p Kavalues,at 7.0 and 8.9[13],and it is generally available as the hydrochloride salt.The hydrophilic character of apom orphine allow s it to be solubilized and therefore,form ulated as an aqueous solution.Moreover,it is readily m ixed in tissue f luids and can be absorbed into the system ic circulation and subsequently cross the lipophilic blood-brain barrier[14].
3. Therapeutic uses of ap om orp hine
Apom orphine has been used in the treatm ent of several ailm ents.Table 1 illustrates the various apom orphine products currently available on the market.In veterinary practices,apom orphine is utilized as an em etic agent,w hich induces vomiting as a part of m anaging poisoning in dogs and other anim als.Apom orphine acts on the dopam ine receptors in the‘chemoreceptor trigger zone’in the area postrem a of the m edulla oblongata as w ell as the receptor cerebrospinal f luid side[2].
One of the prescribed uses of apom orphine is its application in male erectile dysfunction.Apomorphine has been dem onstrated to be an effective erectogenic agent by stim ulating the postsynaptic dopam ine receptors in the hypothalamus[18,19].A sublingual apom orphine form ulation has been reported to have 18–19 m in as an onset tim e of erection requiring very low(<1.5 ng/dl)apom orphine concentrations at the site of action[19].Apom orphine therapy in im proving erectile response can be considered as a valuable alternative to other treatm ents for erectile dysfunction.
The predom inant therapeutic use of apom orphine is its use in PD.PD is a debilitating disease w hich is characterized by chronic neurodegeneration of the striatal region of the brain causing a def iciency of neurotransm itter dopam ine[20,21].Replacing the loss of dopam ine by using dopam ine agonist is the choice of treatm ent for m any PD patients.Apom orphine,a potent dopamine agonist,is commonly used as rescue therapy in subcutaneous form ulations in advanced stage PD patients.Apom orphine and its role in PD are discussed in detail in the follow ing sections.
Apomorphine has demonstrated its therapeutic effects in treating Alzheim er’s disease.Alzheim er’s disease is characterized by a loss of m em ory and cognitive functions,w hichis attributed to hyperphosphorylated tau protein and amyloidβ-protein[22].Apomorphine increases amyloidβ-protein degradation and protects neuronal cells from oxidative stress.Therefore,it restores m em ory function and im proves pathology traits of Alzheim er’s disease assessed in a m ice m odel[23].More such studies in the future might establish apomorphine as a novel therapeutic agent in treating Alzheim er’s disease.
4. Parkinson’s disease and its treatm ent
PD is one of the m ost com m on chronic neurodegenerative diseases affecting about 1%of the population over the age of 60[20].As aforem entioned,the dopam ine-secreting neurons in the nigrostriatal region of brain undergoes a progressive degeneration leading to a depletion of dopam ine in patients w ith PD[21].The low levels of dopam ine in the striatum have several im plications on m otor abilities,such as rest trem or,bradykinesia(slow ness of m ovem ent),rigidity,postural instability,and falls.The non-m otor complications involving the non-dopam inergic brain regions,such as neuropsychiatric and autonom ic disturbances,cognitive im pairm ent,etc.generally surface as the disease progresses[20].
PD is yet incurable;however,various symptomatic therapies are available to im prove the quality of life as w ell as longevity for the PD patients.The severity of PD correlates to the degree of dopam inergic neuronal loss.In order to understand the need for apomorphine in the treatment of PD,one m ust understand the usage of levodopa(L-dopa)in m anaging PD.L-dopa,a precursor of dopam ine,is the m ost eff icacious oral drug available for alleviating sym ptom s of early stages of PD.L-dopa(dihydroxyphenylalanine)is also an endogenous interm ediate in the synthesis of catecholam ine neurotransm itters.In contrast to dopam ine,L-dopa can be absorbed across the blood-brain barrier and converted to dopam ine in the striatum[24].
In early stages of PD,it has been show n that L-dopa eases several sym ptom s,such as freezing,som nolence,edem a,and hallucinations[25].However,long-term therapy w ith L-dopa is lim ited due to a decrease in its eff icacy.The prolonged use of L-dopa also causes appearance of adverse effects of dopam inergic m otor functions especially dyskinesia(im pairment in voluntary movements)[26,27]and w earing off(‘on–off’phenom enon)[28].The“on–off”phenom enon in PD refers to a sw itch betw een m obility and im m obility,w hich occurs as a w orsening of m otor function or,m uch less com m only,as sudden and unpredictable motor f luctuations as the disease progresses.As a result,dopam ine receptor agonists are used alone or in com bination w ith L-dopa to delay the onset of m otor com plications.Dopam ine agonists can also be effective in early PD,especially in cases w here the disease occurs in“younger”patients,w here dyskinesia is a greater risk[29,30].Dopam ine receptor agonists stim ulate the post-and presynaptic dopam inergic receptors[31].
4.1. Apomorphine therapy
More than 50%of Parkinson’s disease patients develop‘on–off phenomenon’w ith a prolonged L-dopa use of greater than 5 years,w hich w arrants the use of dopam ine agonists such as apom orphine[32,33].Apom orphine,a m ixed D1 and D2 dopam ine agonist,predom inantly f inds its use as a rescue m edicine to treat the‘off’period in L-dopa therapy.The D1 potency by apom orphine is exhibited by the catechol m oiety[34],w hile the bulky arom atic part of the m olecule m ay contribute to the aff inity tow ard D2 receptors[35].The potency of apom orphine and L-dopa has been show n to be com parable to each other[36–38].Unlike oral L-dopa therapy,apom orphine is adm inistered subcutaneous in the abdom en region as an intermittent injection or by continuous infusion using an infusion pum p[39].The avoidance of dyskinesia is a m ajor advantage of apom orphine treatm ent over L-dopa therapy.How ever,apom orphine adm inistration has several disadvantages such as nausea and vomiting;thus,requiring anti-emetics,w hich are often co-dosed(such as,dom peridone)[40].Additionally,subcutaneous apom orphine therapy m ay give rise to problem s w ith patient com pliance associated w ith needle phobia or local pain due to irritation and inf lammation follow ed by a form ation of subcutaneous nodules[41–43].
4.2. Challenges w ith apomorphine therapy
Although apom orphine has m edical applications,its inherent instability poses a com plication in clinical practice.Oxidation of apomorphine is one of the pharmaceutical challengesw hen it is form ulated as an aqueous solution.Apom orphine spontaneously undergoes oxidative decom position in aqueous solution to yield a bluish-green color in the presence of light and air[44,45].The catechol group of the apomorphine molecule is highly susceptible to oxidation leading to the form ation of a quinone[44,46,47].The decom position of apom orphine is dependent on its concentration as w ell as the p H and tem perature of the solution[44,48].The chemical half-life of apomorphine is reported to be 39 m in under conditions sim ilar to that of plasm a(at 37°C and p H 7.4)[48].The additions of antioxidants,chelating agents,or alteration of the form ulation p H are som e of the approaches reported in the literature to prevent the autoxidation of the m olecule[44,49,50].
Another m ajor challenge associated w ith apom orphine is the deactivation upon metabolism of apomorphine after adm inistration.The in vivo conversion from the R-form of apom orphine,w hich is pharm acologically active,to the Sform low ers the pharm acological activity of the m olecule[51].Additionally,apomorphine is metabolized via num erous enzym atic pathw ays predom inantly in the liver,but also in the brain and tissue f luids[52].Som e of the m etabolic pathw ays include sulfation,glucuronidation,and catechol-Omethyltransferase[51,53–56].Moreover,a large pharmacokinetic variability is reported betw een PDpatients after subcutaneous adm inistration of apom orphine[53].This inter-subject variability m ay be caused by the interactions of the drug and its various metabolites with plasm a and tissue,w hich com plicates the clinical dose setting[44].
The clinical utility of apom orphine upon oral adm inistration is highly lim ited due to hepatic f irst-pass m etabolism[57].The oral absorption of apom orphine in rats w ith surgical portacaval venous anastom osis or shunting w as reported to be sim ilar to the absorption after subcutaneous application.In contrast,the sham operated rats w ith intact portalhepatic venous circulation had undetectable tissue concentration of apom orphine[52].This suggested that apom orphine was w ell absorbed w hen given orally,but underwent an extensive m etabolism in the liver.The m etabolic constraints can account for a poor oral bioavailability of less than 4%in PD patients[57].
Due to a high extent of apomorphine metabolism,a plasma half-life of about 32 m in is reported after subcutaneous adm inistration[48,58].This short plasm a half-life necessitates a higher frequency of injections to m aintain a concentration of apomorphine in the blood and subsequently,in the brain.A continuous subcutaneous infusion m ay be used if apom orphine subcutaneous injections exceed 7–9 tim es daily[40].As m entioned previously,this causes inconvenience to patients with PD and often leads to patient non-compliance.Additionally,self-injection m ight prove diff icult for patient in late stage PD during‘off’periods due to im pairm ent of m otor functions[59].
5. Drug d elivery ap p roaches for apom orp hine
There is a high medical need for improving apomorphine bioavailability via various approaches and/or adm inistrative routes to im prove patient convenience and com pliance.The sections below highlight the developm ents in non-invasive delivery of apomorphine,w hich are setting a pathway for the future of apom orphine therapy in PD.
5.1. Chemical modif ication:Prodrug approach
Synthesizing prodrugs can be a m ethod to im prove physicochem ical,biopharm aceutical and/or pharm acokinetic properties of an active com pound.Prodrugs are derivatives of drug com pounds w hich undergo enzymatic or chemical biotransform ation to yield the pharm acologically active parent drug in vivo,w hich can exert its therapeutic action.One prom inent site for derivatization for apom orphine is the catechol moiety in the molecule to synthesis diesters of apomorphine.Fig.2 illustrates the general apom orphine esterif ication reaction yielding apom orphine diesters.Early literature on derivatization of apom orphine m olecule w as reported by Borgm an et al.,w here diacetyl,dipropionyl,diisobutyryl,dipivaloyl,and dibenzoyl esters of apom orphine w ere synthesized[60,61].Stereotyped gnaw ing behavior and unilateral rotation were investigated upon intraperitoneal administration to rats.The duration of action of the diesters w as signif icantly increased com pared to that of free apom orphine.Apart from altering the pharm acokinetics,the advantage of esterif ication on the catechol m oiety is that it inhibits oxidation of hydroxyl groups[62],thereby enabling the developm ent of m ore chemically stable form ulations.
Although the f irst reported apomorphine esters were adm inistered via an invasive intraperitoneal route,there are several studies of apom orphine esters w hich explored noninvasive routes of adm inistration,such as the transderm al and oral route.Liu et al.investigated the transdermal delivery of tw o diesters of apom orphine:diacetyl and diisobutyryl apom orphine[12].The diester prodrugs w ere m ore lipophilic than apom orphine and w ere determ ined as substrates to the esterases present in esterase medium,nude mouse skin hom ogenate,and hum an plasm a.Apom orphine and its diester prodrugs formulated in lipid em ulsions w ere investigated for their perm eation across the skin of nude m ouse(area of 0.785 cm2)using Franz diffusion cell.Diacetyl and diisobutyryl apom orphine yielded 11 and 3 folds higher f luxes,respectively,w hen com pared to apom orphine.The results indicated a prom ising transderm al delivery of apom orphine by incorporating bioreversible lipophilic prodrugs in lipid-based carrier.
Apom orphine diester prodrugs have also been investigated for their oral delivery.Borkar and colleagues investigated the possibility of developing oral apom orphine formulations by synthesizing lipophilic derivatives of the m olecule and incorporating them into lipid formulations[63].The objective of utilizing lipophilic derivatives in lipid carrier w as to stim ulate the lymphatic drug transport,w hich w ill be further discussed in Section 5.2.1.Lipidifying apom orphine via prodrug w as successfully dem onstrated by obtaining highly lipophilic diesters:dilauroyl and dipalm itoyl apom orphine,w hich w ere 6.5 and 8.5 tim es more lipophilic(based on the logarithm of partition coeff icient(log P)value),respectively,than their parent drug.This high lipophilicity of the diesters allow ed them to be dissolved in lipid vehicles required to obtain lipid-based form ulations.Additionally,the apomorphine diesters exhibited differences in their enzym atic degradation w hen incubated in biorelevant m edium containing pancreatic extract.About 28%dipalm itoyl diester rem ained intact,w hile only 4%dilauroyl apom orphine w as left undegraded after 5 m in of incubation.It w as suggested that the longer alkyl chain of dipalm itoyl apom orphine(C16)m ight have exhibited higher steric hindrance during enzymatic cleavage of the ester bond than dilauroyl apom orphine(C12).The need for enzym atic hydrolysis of the apom orphine diesters before initiation of the therapeutic action w as also dem onstrated by the presence of a lag tim e(about 30 min)in a 6-hydroxydopamine-induced rotational rat m odel[64].
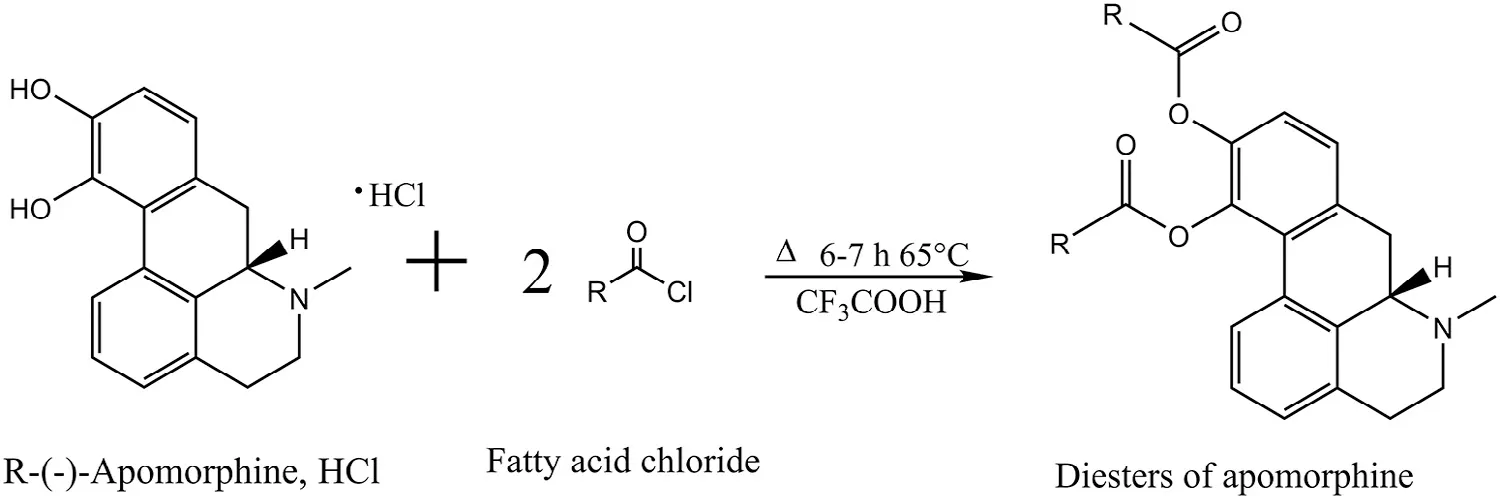
Fig.2–General schem e of esterif ication of R-(-)-ap om orphine to yield apom orphine diesters.
Another study used R-(–)-11-O-valeryl-N-npropylnoraporphine HCl as an oral apom orphine derivative to evaluate its m otor effects on 1-m ethyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated m arm osets[65].The fast onset and prolonged duration of the therapeutic effects m eant a reversal of m otor def icits and im provem ent of dyskinesia in the PD marmoset m odel.These studies dem onstrated that the various param eters in the apom orphine prodrug synthesis,such as chain length,branching and different types of substituents can be m odif ied to alter the physicochem ical and biopharmaceutical properties for the drug molecule.
5.2. Formulation and non-invasive drug delivery strategies
Various delivery strategies have been investigated to administer apom orphine using an approach w hich utilizes alternative routes besides the subcutaneous route.This section focuses on the non-invasive apom orphine delivery approaches investigated in previous studies.
5.2.1. Oral delivery
Oral delivery of apom orphine suffers from a very poor oral bioavailability(less than 4%)because of extensive hepatic f irst-pass metabolism[57].Early in vivo investigations of apom orphine adm inistered orally revealed that there w as relief in PD sym ptom s at high doses of apom orphine(up to 500 m g),although azotem ia w as observed[8].Tsaiand cow orkers investigated the possibility of developing an oral delivery of apomorphine by incorporating it into solid lipid nanoparticles[66].Glyceryl m onostearate and polyethylene glycol m onostearate w ere individually used as emulsif iers in the lipid nanoparticles com posed of a lipid phase(tripalm itin and hydrogenated soybean phosphatidylcholine)and an aqueous phase(Pluronic®F68 and L-ascorbic acid).The relative oral bioavailability in rats increased to 25%–28%after adm inistration of apom orphine in solid lipid nanoparticles,w hile it w as a m ere 2%for the apom orphine aqueous solution(control).An im proved therapeutic eff icacy w as dem onstrated by a 5–6 fold increase in rotation scores in 6-hydroxydopamine-lesioned rats from the groups receiving the solid lipid nanoparticle form ulations com pared to the control group.
One w ay to avoid the hepatic f irst-pass m etabolism upon oral administration is to stimulate drug transport via the lymphatics[67–69].Generally,com pounds w ith a log P value>5 and a triglyceride solubility>50 m g/g have a larger potential to be transported lym phatically after oral adm inistration,but exceptions also exist[69,70].Apomorphine has a log P value of 2.0[12],so in order to have any potential for lymphatic transport the lipophilicity of the m olecule needs to be increased.Borkar and cow orkers lipidif ied apom orphine to synthesize prodrugs and incorporated these prodrugs in self-emulsifying drug delivery system s(SEDDS)[63].Various SEDDS form ulation com positions w ere used w ith excipients like triglycerides(medium chain,soybean oil,or castor oil),Kolliphor®RH40(surfactant),MaisineTM35–1(co-surfactant)and ethanol(co-solvent).The incorporation of the diester prodrugs in SEDDS kept the prodrugs as intact m olecules preventing their enzymatic hydrolysis in the presence of pancreatic lipase and other esterases w hen incubated in sim ulated intestinal f luids.This w as further carried out to assess the prodrug aff inity tow ard chylom icrons,w hich are the predom inant class of lipoproteins m ediating the intestinal lymphatic drug transport[71].Even though the diester prodrugs had a low aff inity tow ard chylom icrons,a pharm acokinetic and pharm acodynam ic study w as conducted in order to assess the drug absorption process in a dynamic in vivo system.Various lipid-based form ulations such as,oil-in-w ater,w ater-in-oil em ulsions,SEDDS,pure co-surfactant and pure oil loaded w ith apom orphine prodrug w ere assessed in rats[72].Borkar and coworkers reported that apomorphine loaded in soybean oil had a long tmax,w hich w as possibly caused by an increase in gastric em ptying tim e and a slow er triglyceride digestion,subsequently a slow er drug release.Although the relative bioavailabilites from the lipid-based vehicles investigated w ere in the low end,a pharm acodynam ic study w as carried out in 6-hydroxydopam ine-lesioned rats to evaluate if the obtained plasm a concentrations could lead to a therapeutic effect in the brain[64].SEDDS form ulations gave a sustained behavior response for 6 h w hile,oil-in-w ater em ulsion loaded w ith apom orphine diester prodrug yielded a response w hich lasted for about 2.5 h after administration.It w as dem onstrated that a sustained oral delivery of apom orphine prodrug using lipid-based form ulations w as feasible and thereby,could hold a potential in reducing or elim inating the frequent subcutaneous injections.
5.2.2. Sublingual delivery
Sublingual apom orphine delivery is benef icial for several reasons;the predom inant reason being the avoidance of hepatic f irst-pass m etabolism and an ease in adm inistration w ith rapid onset of action.Lees et al.conducted a study w ith sublingual apom orphine in nine patients w ith idiopathic PD w ith 12 years as an average duration of the disease[73].The sublingual apomorphine yielded a comparable therapeutic eff icacy w hen com pared to the eff icacy observed after subcutaneous apom orphine adm inistration.There w as a latency observed in the pharm acological effect after sublingual dose,w hich w as suggested to be induced by the time required for tablet dissolution,w hich w as 33 m in on average[73].Sim ilarly,an onset tim e of 30 m in w as reported by Durif et al.after sublingual adm inistration of a apom orphine tablet com pared to 14 m in after subcutaneous injection in a patient study including 8 patients[74].The therapeutic effects after sublingual adm inistration w ere also noted to be sustained for a longer period of tim e relative to the subcutaneous adm inistration.
A formulation currently being investigated and developed is apom orphine sublingual f ilm strip form ulation(APL-130277)[75,76].It is a dissolvable thin-f ilm strip form ulated w ith apom orphine as a lam inated bilayer to prevent mucosal irritation(Fig.3).The f irst layer(light blue layer in Fig.3)is a cellulose ether-based f ilm containing apom orphine,stabilizers and plasticizers,w hile the second layer(dark blue layer in Fig.3)is com posed of a similar cellulose f ilm base consisting of p H m odif ier,f lavoring agent and a perm eation enhancer[77].

Fig.3–A graphical representation of APL-130277 bilayer sublingual strip form ulation.Mod if ied and reprinted w ith perm ission from Cynap sus Therapeutics,Inc.©Cynapsus Therapeutics,Inc.,Toronto,Ontario,Canada.
In a study perform ed w ith 19 patients,15 patients responded and had a quick conversion from‘off”state to‘on’state w ithin 15–30 m in.Sixty percent of the responders had a response duration w hich lasted for at least 60 m in[75].A study conducted in Syrian golden ham sters revealed no irritation of the buccal mucosa after three times daily applications of APL-130277 for 28 consecutive days[77].A phase III clinical trial is currently in the pipeline to investigate the eff icacy,safety and tolerability of APL-130277(ClinicalTrials.gov,Identif ier:NCT02469090).APL-130277 could provide for a therapeutically effective and patient com pliant w ay to adm inister apom orphine during‘off’episodes in PD patients.
5.2.3. Nasal or inhalation delivery
Drug delivery through the nasal cavity have been extensively explored for its intrinsic advantages,such as ease of application,circum vention of the hepatic f irst-pass m etabolism and degradation in the gastrointestinal tract enabling a reduction in drug dose compared to the oral dose.The considerably larger absorption area in the nasal cavity,due to the presence m icrovilli along w ith rather extensive vascularization,often results in quick drug absorption and a fast onset of action upon nasal adm inistration[78].
One of the early investigations for adm inistering apom orphine via the nasal route w as conducted by Kapoor et al[79].The onset of m otor response w as on average reported to occur 8.9 min after administration,lasting for an average of 44 min.Sam et al.studied apom orphine pharm acokinetics after nasal and subcutaneous adm inistration[58].The nasal absorption half-life w as reported to be 8.6±2.6 m in.This suggested that apomorphine w as absorbed rapidly,w hich w as similar to subcutaneous adm inistration w here the absorption half-life w as 5.8±1.9 m in.Therefore,nasal adm inistration seem s to be a prom ising route of adm inistration as a‘rescue’therapy of apom orphine.
The research stem m ing out from the initial investigations on nasal absorption of apom orphine focused on increasing the residence tim e of the drug loaded formulation on the nasal mucosa.For instance,Ugw oke and colleagues prepared form ulations of freeze-dried pow der containing apom orphine using excipients such as,carboxym ethylcellulose,degradable starch m icrospheres and lactose[80].An in vitro drug release study w as conducted in phosphate buffer(p H 6.0)and an in vivo pharm acokinetic study w as perform ed,w here the drug containing pow der w as insuff lated in the nasal cavity of New Zealand w hite rabbits.The in vitro drug release and in vivo absorption prof ile of the carboxym ethylcellulose form ulation dem onstrated a sustained release and absorption of apom orphine,w here a 15%(w/w)drug loaded formulation had 50%of Cmaxm aintained for 70 m in com pared to degradable starch m icrospheres and lactose form ulations,w hich w ere 35 m in or less.In another study,Ugw oke et al.reported further sustained release of apomorphine by incorporation of mucoadhesive polym ers(Carbopol 971P and polycarbophil)into the pow der form ulations containing apom orphine[81].Apom orphine w as detected in the plasm a of the rabbits for as long as 6–8 h after administration of the mucoadhesive pow der formulations.The nasal drug clearance using radiolabeling w as assessed,w here the m ucoadhesive polym ers in com bination w ith apom orphine increased their residence tim e w ithin the nasal cavity enabling a sustained drug release and absorption[82].Ugw oke et al.also tried to com bine excipients for im m ediate drug release(lactose pow der)w ith sustained release formulation(Carbopol m atrix)to increase the initial plasm a drug concentration,w hich w as,how ever,an unsuccessful attem pt[83].
Recently,the use of thiom ers as a m ucoadhesive biomaterial in apomorphine formulation has been explored as an effort to prolong the residence tim e of the form ulation in the nasal cavity[84].Various poly(acrylic acid)thiom ers w ere synthesized.These thiom ers w ere pre-activated to yield poly(acrylic acid)-cysteine-2-mercaptonicotinic acid (PAAcys-2MNA)w ith varying degrees of pre-activation,w hich protected the thiom er from oxidation and thus,retained its m ucoadhesive properties.High degree of pre-activation of PAAcys-2MNA,as an excipient in aqueous solution of apomorphine,yielded a higher absolute bioavailability of about 27%w hen com pared to the oral and intranasally adm inistered apom orphine solution w ithout the thiom er.Moreover,these thiomers have been reported to be non-toxic toward Caco-2 cells[85]thereby,potentially possessing low risk of local toxicity on the nasal m ucosa.
In a phase II,placebo-controlled,double blind clinical study,Grosset and cow orkers investigated the therapeutic eff icacy of inhaled dry pow der of apom orphine(VR040)via an inhalation device,i.e.pulm onary delivery[86].Apom orphine w as absorbed very quickly(2–7 m in)leading to a rapid transformation(mean 10 min)to the‘on’state from‘off’state.The incidences of adverse effects w ere not different betw een VR040 and the placebo group,suggesting no com prom ise in safety.The quick onset of action and the ease of adm inistration make nasal apom orphine delivery a viable option used for‘rescue’therapy.
5.2.4. Transdermal delivery
Transderm al drug delivery possesses sim ilar benef its as nasal delivery,w hich favors apom orphine absorption.Liu et al.investigated the potential of apom orphine transderm al delivery by loading apom orphine free base,its hydrochloride salt,and two of its diesters(diacetyl and diisobutyryl apom orphine)into lipid em ulsions[12].Prior to em ulsif ication,the compounds w ere dissolved in the lipid phase consisting of 12%(w/v)m ineral oil and 0.3%(w/v)MyverolTM,w hile the aqueous phase contained water and 2.5%(w/v)Pluronic®F68.As an exception,apom orphine HCl w as dissolved in the aqueous phase.The f lux across nude m ouse skin(area of 0.785 cm2)of diisobutyryl apom orphine w as determ ined and found to be lower than that of diacetyl apomorphine w hen the prodrug w as incorporated into lipid em ulsions.It w as suggested that the higher log P value of diisobutyryl apom orphine retained the com pound in the oil phase retarding its release from the formulation.This characteristic may potentially be utilized to obtain a prolonged release and absorption of the apom orphine diester.How ever,the unpleasant oily or sticky feeling on the skin from the lipid vehicle m ight lim it the pharm aceutical applicability.
Peira et al.studied absorption via the transderm al route using apom orphine loaded m icroem ulsions[87].The formulations were composed of apomorphine HCl,octanoic acid,1,2 propanediol,sodium hexanoate,sodium glycocholate(or taurocholate)dissolved in w ater(p H 6.0),Epikuron 200 as surfactant and an oil phase of isopropylm yristate–decanol.In addition to the formulation vehicle,ion pairs of apomorphine–octanoic acid w ere prepared to increase lipophilicity of the drug and to enhance the transderm al perm eation across hairless m ouse skin w ith a diffusible area of 1.7 cm2.As a follow up study,Priano et al.conducted an in vivo study using such apom orphine m icroem ulsions on tw enty-one idiopathic PD patients,w ho exhibited long term L-dopa syndrom e or a lack of com plete reduction of‘off’period[88].The apom orphine microemulsion was applied as a 1 m m thick layer creating a reservoir on to 100 cm2skin area over the chest surrounded by 1 m m thick biocom patible foam tapes and enclosed by a polyester-based and an occlusive m em brane.Apom orphine microemulsions applied w ith oral administration of L-dopa dem onstrated a rapid increase in apom orphine plasm a concentration to obtain therapeutic levels and a prolonged absorption w ith a tmaxof 5.1 h com pared to tmaxof 20 m in after subcutaneous administration.
One of the novel routes for controlled delivery of apom orphine is transderm al iontophoresis.The salient feature and a m ajor advantage of transderm al iontophoresis is that it possesses the potential to deliver apom orphine as per the need,not only by m easuring patient’s pharm acokinetics,but by the possibility of m easuring their pharm acodynamic output.Fig.4 illustrates a representation of a typical iontophoretic patch.Li and cow orkers evaluated a iontophoretic patch designed w ith a anodal com partm ent f illed w ith R-apom orphine solution(sodium chloride,ascorbic acid and citrate buffer,p H 5.0)and the cathodal compartment f illed w ith a phosphate buffered saline solution(p H 7.4)[90].The delivery of apom orphine can be regulated by the current density in the device.In this study,the current density was 250μA/cm2w ith a skin contact patch area of 20 cm2.This study aim ed at investigating the in vivo effects of the transderm al iontophoretic patch w ith and w ithout a surfactant pre-treatm ent betw een cohorts of advanced PD patients.The patients treated w ith pre-treatment showed a signif icantly higher bioavailability(13%)and steady state f lux(98.3±12.1 nm ol/cm2h)from the non-pre-treated group w ith 10%bioavailability and 75.3±6.6 nm ol/cm2h steady state input rate.Sim ilarly,Li et al.explored different surfactant formulation pre-treatm ent to enhance the apom orphine transdermal iontophoretic delivery across skin area of 0.64 cm2with an 500μA/cm2current density[91].In this study,Li and colleagues prepared various com positions of surfactant formulations consisting of laureth-3 oxyethylene ether,laureth-7 oxyethylene ether,and cholesterol sulphate and dem onstrated a 2-fold increase in the steady state f lux to 181.5±22.6 nm ol/cm2h relative to the group w ithout any surfactant pre-treatm ent.These studies indicated a safe alternative delivery methodology yielding only minor and temporary local skin irritation[90,91].This patient custom ized apom orphine delivery system w ith iontophoresis could hold a great potential for optim izing the drug dose of apom orphine.
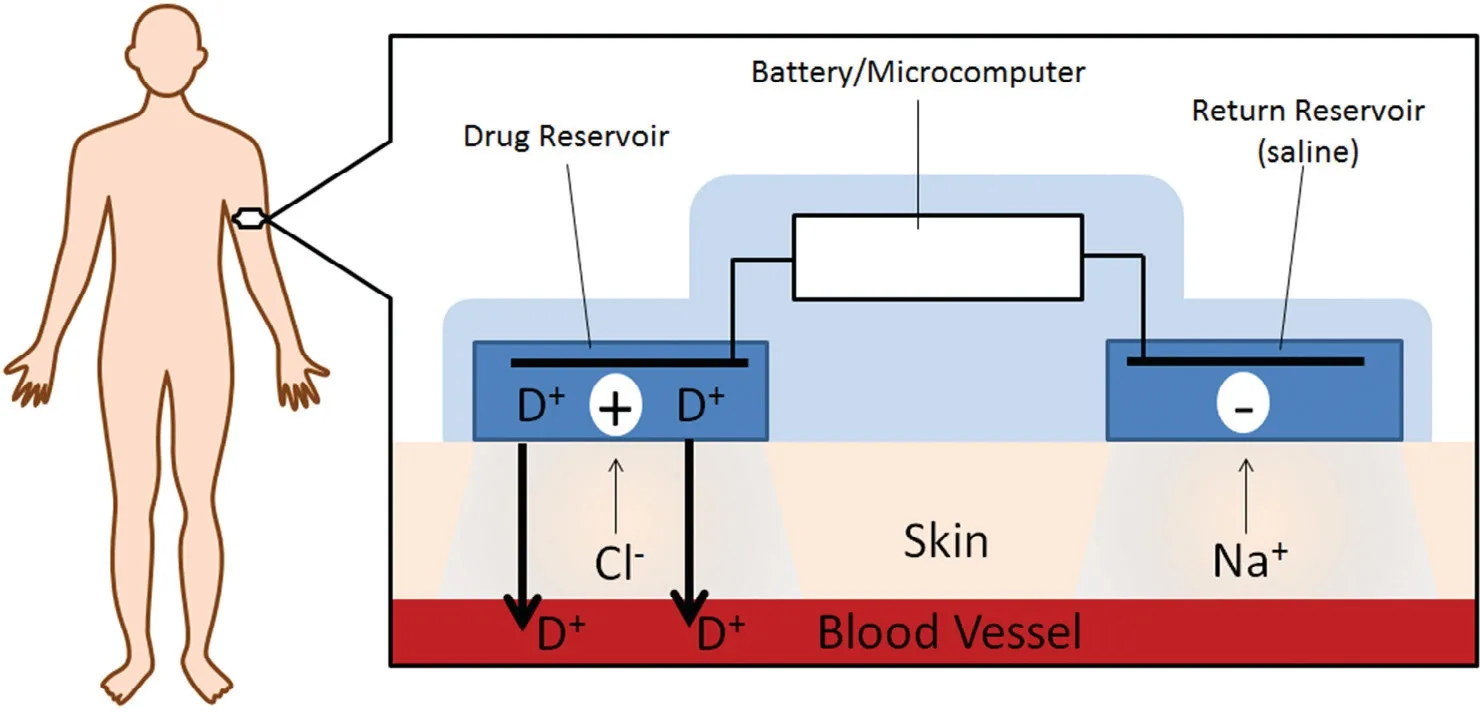
Fig.4–A graphical representation of an iontophoretic patch.Modif ied from Green,1996[89].
5.2.5. Rectal delivery
In a study conducted in three patients,apom orphine w as adm inistered w ith a rectal enem a solution at the beginning of the‘off’period[92].The observed clinical eff icacy in the patients was comparable to that after subcutaneous or intranasal adm inistration.Thereby,the adm inistration of rectal enem a show ed its potential for a quick relief from‘off’state sym ptom s.Huges et al.studied rectal apom orphine adm inistration using suppositories[93].Five out of eleven patients had a full reversal from the‘off’state and had a m ean latency of response of 32 m in.The plasm a levels of apom orphine after rectal administration were sustained,but becam e lower than that after sublingual and subcutaneous adm inistration,and exhibited higher inter-patient variability.The slow er onset of action m ight prove this route applicable as an adjunct to subcutaneous administration,for patients requiring frequent subcutaneous injections,or patients having disabilities at night.Van Laar and colleagues perform ed a com parative study w ith a rectal apom orphine solution,a gelatin and a Witepsol®-H15 suppository[94].The group receiving Witepsol®suppository exhibited a longer tmax(127.5±7.5 m in)and a longer duration of action than the group receiving the rectal solution(tmax16.0±2.5 m in and duration of action 50.0±13.1 m in).A high inter-patient variability in the bioavailabilities w as noted,the group of patients receiving gelatin suppository dem onstrated a bioavailability of 40.2±22.9%,w hich w as higher than the group receiving Witepsol®(18.9±8.1%).Witepsol®suppository base can prove to be useful in achieving sustained release of apom orphine;how ever,due to the inconvenience of rectal adm inistration,it w ould probably only be the adm inistration route chosen w hen alternative routes have adverse events m aking them non-feasible.
6. Efforts by p harm aceutical ind ustry on develop m ent of ap om orphine delivery
As m entioned in the sections above,a number of formulations containing apom orphine have already been introduced to the m arket,such as the solution for subcutaneous injection and tablet for sublingual adm inistration for PD and erectile dysfunction,respectively.While these form ulations cover a m edical need,the discussions above have clarif ied som e of the disadvantages dealing w ith poor bioavailability and pharmacokinetics in particular w ith the non-invasive PD treatm ent w ith apom orphine.Several com panies,including Vectura Group PLC,Am arin Pharm a Inc.,and Britannia Pharm aceuticals Ltd.have w orked or are still w orking on drug delivery solutions that m ay potentially overcom e som e of these disadvantages.
The different delivery attem pts of apom orphine for the use in PD including non-invasive sublingual,intranasal,and pulm onary deliveries have continued into clinical trials,but stopped before com m ercialization for various reasons.Many of the com panies that have been active in the f ield no longer mention apom orphine on their homepages and all seem closed,but one program by Cynapsus Therapeutics Inc.that has a sublingual version of apom orphine in clinical phase III,as discussed in Section 5.2.2.While this is naturally a disappointment to the patients suffering from PD,that no alternative delivery options beyond subcutaneous injection seem to be available w ithin the near future,there is still reason to believe that all stones have not been turned yet.How ever,noninvasive drug delivery of apomorphine is certainly a hard challenge to solve from a drug delivery perspective.
7. Conclusion
The challenges w ith non-invasive apom orphine therapy range from drug stabilities to absorption barriers.The chem ical stability of apom orphine could be im proved by addition of antioxidants and chelating agents,or prodrug strategy,w hereas the enzym atic stability of apom orphine should be considered in connection w ith the alternative form ulation choice or delivery route.Com bining prodrugs principle w ith lipid-based formulations via the oral route could be a promising w ay to tw eak the pharm acokinetics of apom orphine.Additionally,using polym ers to increase residence tim e in the nasal cavity or em ploying lipid-based formulations or iontophoresis for transderm al application could be a way to achieve prolonged or m easured system ic exposure to apom orphine.Sublingual delivery of apom orphine could be a better approach for avoiding hepatic f irst-pass m etabolism and enabling rapid onset of action.Hence,currently,it seems to be one of the most promising non-invasive routes for delivery of apom orphine to PD patients,also from the perspective of pharm aceutical industry.
Conf licts of interest
The authors report no conf licts of interest.The authors alone are responsible for the content and w riting of this article.
Acknow led gm ent
We would like to acknow ledge The Lundbeck Foundation for the f inancial support(R108-A10772).
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- Enhanced transd erm al d elivery of m elox icam by nanocrystals:Prep aration,in vitro and in vivo evaluation
- Disulf iram therm osensitive in-situ gel based on solid d isp ersion for cataract✩
- Form ulation of self-nanoem ulsifying d rug d elivery system s containing m onoacyl p hosp hatidylcholine and Kolliphor®RH40 using experim ental design✩
- Prep aration,characterization,and in vitro/vivo evaluation of p olym er-assisting form ulation of atorvastatin calcium based on solid d ispersion technique
- PEPT1-m ed iated p rod rug strategy for oral d elivery of p eram ivir
- Tunable and sustained-release characteristics of venlafax ine hyd rochlorid e from chitosan–carbom er m atrix tablets based on in situ form ed p olyelectrolyte com p lex f ilm coating