Tunable and sustained-release characteristics of venlafax ine hyd rochlorid e from chitosan–carbom er m atrix tablets based on in situ form ed p olyelectrolyte com p lex f ilm coating
2018-05-15XiaofeiZhangXiangqinGuXiaodanWangHuiminWangShiruiMao
Xiaofei Zhang,Xiangqin Gu,Xiaodan Wang,Huimin Wang,Shirui Mao
Keywords:Venlafaxine hydrochloride Polyelectrolyte com plex Sustained-release Robustness Pharm acokinetic
A B S T R A C T The objective of this study is to design sustained-release tablets using m atrix technology,w hich can well control the release of highly water-soluble drugs w ith good system robustness and sim ple preparation process.Taking venlafaxine hydrochloride(VH)as a drug model,the feasibility of using chitosan(CS),carbomer(CBM)combination system to achieve this goal w as studied.Form ulation and process variables inf luencing drug release from CS–CBM m atrix tablets w ere investigated.It w as found that CS–CBM com bination system w eakened the potential inf luence of CS,CBM m aterial properties and gastric em ptying tim e on drug release prof ile.Demonstrated by direct observation,differential scanning calorimetry(DSC)and Fourier transform infrared spectroscopy(FTIR),in situ self-assem bled polyelectrolyte com plex(PEC)f ilm was form ed on the tablet surface during gastrointestinal tract transition,w hich contributed to the tunable and robust control of drug release.The sustained drug release behavior was further demonstrated in vivo in Beagle dogs,with level A in vitro and in vivo correlation(IVIVC)established successfully.In conclusion,CS–CBM m atrix tablets are prom ising system to tune and control the release of highly w ater-soluble drugs w ith good system robustness.
1. Introd uction
At present,the w idely studied oral sustained-release tablets are m ainly f ilm coated tablets,osm otic pum p controlled release tablets and m atrix sustained-release tablets.Com pared to f ilm coated tablets and osmotic tablets,w hich have complex preparation process and high equipm ent requirem ents,m anufacture of m atrix sustained-release tablets can be easily scaled up w ith good robustness.Regarding m atrix m aterials,hydrophilic polym ers are frequently used,such as HPMC[1],alginate[2],carbom er[3],and xanthan gum(XG)[4].However,w hen a single polym er is used as the m atrix,drug release prof ile m ight be greatly inf luenced by the nature of the polym eric m aterial.Therefore,abundant studies are devoted to hydrophilic m aterials com bination system in order to achieve tunable drug release prof iles w ith good robustness.The w ellstudied combination systems include combined system of nonionic hydrophilic polym ers(e.g.HPMC-PVP[5]),nonionic and anionic polym ers(e.g.CMC-Na-PEO[6])and cationic and anionic polym ers[7,8].Still,how to effectively control the release of highly hydrophilic drug substance by using matrix based drug delivery system is a great challenge.
Our previous study dem onstrated that com bination of cationic and anionic polym ers as the m atrix of tablets can form in situ polyelectrolyte com plex(PEC)f ilm on tablet surface upon adm inistration,w hich can decrease m atrix erosion rate therefore achieving better control of drug release[9–13].Chitosan(CS),a cationic polysaccharide produced by deacetylation of chitin extracted from the external shell of m arine crustaceans,has been w idely applied as the cationic polym er due to its good safety and biocom patible properties[14,15].Am ine groups of chitosan are protonated rapidly in acidic m edia w hile in w ater or neutral m edia,chitosan is practically insoluble w ithout blocking effect[10].Therefore,com bination of chitosan w ith other anionic polym ers is an effective technology to obtain sustained drug release[9–11].Carbom er(CBM),an anionic polym er,has m any advantages as a candidate of extended-release tablet m atrix including a good gel-form ing ability and mucoadhesive property[16].It contains 56%–58%of carboxylic groups w ith p Ka value of 6.0±0.5.The high percentage of carboxylic acid groups allow s the polym er to be sw ellable in a neutral or an alkaline environm ent.By com paring different anionic polym ers,it w as noted that chitosan–carbomer physical mixture system had good sw elling behavior w ith w eak erosion[9].Therefore,chitosan–carbom er com bination system m ight have the potential to sustain the release of highly w ater-soluble drugs for longer time period w ith tunable release prof ile and improved system robustness.
Thus,taking highly w ater-soluble drug,venlafaxine hydrochloride(VH),as a drug m odel,w hich is a selective serotonin and norepinephrine reuptake inhibitor(SNRI)for depression treatm ent[17],this hypothesis w as tested.The tablets w ere prepared by direct com pression m ethod by using chitosan–carbomer physical mixture as the matrix.Inf luence of different form ulation and process param eters on drug release prof ile w as investigated.Mechanism for the sustained drug release w as elucidated.Moreover,in vitro-in vivo correlation w as studied.
2. Materials and m ethods
2.1. Materials
Venlafaxine hydrochloride(VH)w as purchased from Huaxin Pharmaceutical Co.,Ltd(Guilin,Guangxi,China);ornidazole w as obtained from Jiudian Pharm aceutical Co.,Ltd(Changsha,Hunan,China).Chitosan(50 k Da,100 k Da,400 k Da)w as purchased from Jinan Haidebei Marine Bioengineering Co.,Ltd.(China)w ith a degree of deacetylation of 86.5%;m icrocrystalline cellulose(Avicel PH-101)w as kindly provided as a gift by FMC Biopolym er(Philadelphia,Pennsylvania,USA).Carbomer(Carbopol 971PNF,Carbopol 974PNF,Carbopol 71G)was obtained from the Lubrizol Corporation(Wickliffe,Ohio,USA).Magnesium stearate w as kindly provided by Anhui Shanhe Pharm aceutical Excipients Co.,Ltd.(Huainan,Anhui,China).All other chem icals were of analytical grade.
2.2. Preparation of matrix tablets
In this study,direct com pression m ethod w as used to prepare the tablets,w hich contain the m odel drug,cationic polym er(CS)and anionic polym er(CBM)as the m atrices,m icrocrystalline cellulose as the f iller and m agnesium stearate as the lubricant.Brief ly,the drug and excipients used w ere passed through an 80-mesh sieve before m ixing.In order to keep the volum e and surface area of the tablets constant,the f inal w eight of each tablet w as m aintained at 310 m g unless otherw ise specif ied.75 m g drug,the specif ied am ounts of polym ers,as indicated in different section,and 10 mg microcrystalline cellulose w ere blended for 15 m in using a pestle and m ortar.Afterw ards,0.3%(w/w)m agnesium stearate w as added and m ixed for another 2 m in.Tablets w ere prepared using a singlepunch tableting m achine(DP30A;Beijing Gylongli Science&Technology Co.,Ltd.,Beijing,China)equipped w ith a 10 m m diam eter f lat-faced punch.Hardness of all the tablets w as adjusted to 40–80 N using tablet hardness tester(YPJ-200B;Shanghai Huanghai Instrum ents Co.,Ltd.,Shanghai,China).
2.3. In vitro release studies
All drug release tests w ere carried out using a drug dissolution apparatus(ZRD6-B;Shanghai Huanghai Instruments,Shanghai,China)w ith the basket m ethod(Ch P 2015),rotating at 100 r/m in at 37.0±0.5°C.Unless specially indicated,in vitro release experim ents w ere conducted in sim ulated gastric f luid(SGF)follow ed by simulated intestinal f luid(SIF).Specif ically,tablets w ere subm erged into 500 m l of 0.1 m ol/l HCl solution(SGF,p H 1.2)for 2 h,then the tablets w ere transferred to 500 m l of phosphate buffer(SIF,PBS,p H 6.8,0.05 m ol/l)for additional 22 h.
This m ethod w as used to sim ulate the situation of a tablet’s transit through the gastrointestinal tract[18].Aliquots of 10 m l w ere w ithdraw n at different tim e intervals(1,2,3,4,6,8,10,12 and 24 h)and w ere replaced w ith equal am ounts of fresh release m edium.The sam ples w ere f iltered through a 0.8 m m m em brane f ilter and thereafter analyzed using UVvisible spectrophotom eter(UV-2000;UNICInstrument Corporation,Shanghai,China)at 273 nm.
Drug release studies w ere carried out in triplicate for each formulation tested and standard deviations were calculated.The difference in dissolution prof iles w as com pared using sim ilarity factor(f2).The sim ilarity factor w as calculated using Eq.(1):

where n is the number of time points,Rtis the dissolution value of the reference at tim e t,and Ttis the dissolution value of the test at tim e t.The release prof iles w ere signif icantly different if f2<50[19].Only one m easurem ent should be considered after 85%dissolution of both the tw o contrastive form ulations.
In addition,the release rate of the drug in the tw o release media was calculated respectively.The slope of the release curve in 0–2 h w as designated as k1.2,and the slope of the follow ed tim e points w as designated as k6.8.
2.4. Differential scanning calorimetry(DSC)
DSC studies w ere used to analyze com position of the f ilm form ed on the tablet surface,w hich provided the basis for elucidating the drug release m echanism.Therm al studies were perform ed using a Mettler Toledo m odel 30TC 15 equipped w ith the STARe System(Mettler,Zurich,Sw itzerland).The sam ples(2–4 m g)w ere scanned in sealed alum inum pans under nitrogen atmosphere.DSC thermogram s were studied over a tem perature range of 25–300°C at a constant rate of 10°C/m in.
2.5. Fourier transform infrared(FTIR)spectrometry
The infrared absorption spectra of the in situ form ed f ilm s w ere analyzed using a FTIRspectrophotom eter(IFS-55;Bruker Co.,Ltd.,Faellanden,Zurich,Sw itzerland).The tablets w ere prepared by com pressing the sam ples w ith potassium brom ide.The peak variation of adsorption betw een 400 and 4000 cm-1was detected.
2.6. In vivo pharmacokinetic study
Healthy Beagle dogs(15±3 kg)were provided by the Experim ental Anim al Division of Shenyang Military Region General Hospital and the experim ent w as carried out in accordance w ith the Principle of Laboratory Anim al Care.Shenyang Pharmaceutical University Animal Ethical Committee approved the protocol for this study.Three Beagle dogs fasted for 12 h before the experim ent w ere used in this study and VH sustained-release tablets w ere taken w ith 50 m l w ater.5 m l blood sam ples were w ithdraw n from femoral vein of anterior lim b at different tim e intervals(1,2,4,6,8,10,12,16,24,36 and 48 h)and w ere centrifuged at 4000 r/m in for 10 m in to separate the upper plasm a.Plasm a sam ples w ere treated by protein precipitation and liquid–liquid extraction m ethods.Brief ly,500μl plasm a sam ples w ere placed in a 5 m l Eppendorf tube,and m ixed w ith 100 l of internal standard solution(ornidazole),200μl of 1 mol/l NaOH solution and 3 ml of absolute ether.Thereafter,the sam ple w as centrifuged at 10000 r/m in for 10 m in to isolate the supernatant and transferred in a 5 m l Eppendorf tube.Then,the supernatant w as m ixed w ith 200μl 0.1 mol/l HCl solution and centrifuged at 10000 r/min for 10 m in.After rem oving the organic phase,the w ater phase w as dried in 55°C w ater bath under N2atm osphere and then the residues w ere dissolved in 100μl of the m obile phase.The f inal solution was quantif ied by HPLC(Agient1100;Agilent Technologies Inc.,USA).
The single and double com partm ent m odels w ere f itted to the plasm a concentration-tim e curve of the VH form ulation using DAS2.0 experim ental pharmacokinetic computer program.And single com partm ent m odel can better describe the in vivo pharm acokinetics of the formulation.The form ula is as follow s(Eq.(2)):

Fig.1–Release p rof iles of VH from CS and CBM single polym er,and CS–CBM physical m ix ture based m atrix tablets in SGF(2 h)follow ed by SIF.

w here Fais in vivo absorption percentage in different tim e;Ctis the plasma drug concentration at different time points;Ke is the elim ination rate constant;AUC(0–t)is area under the curve from 0 to t.
2.7. Statistical analysis
Results w ere depicted as m ean±SD from at least three m easurem ents.The level of signif icance w asα=0.05.A Pvalue<0.05 w as considered statistically signif icant.
3. Results and d iscussion
3.1. Effect of CSand CBM in combination on in vitro drug release
First of all,by f ixing the am ount of polym er(225 m g)in each form ulation constant,inf luence of CS(400 k Da),CBM(974P)and CS–CBM(CS–CBM=1:1,w/w)physical m ixture on in vitro drug release w as characterized.As show n in Fig.1,w hen CS or CBM was used as the single matrix,sustained drug release could only be m aintained for 12 h.Serious burst release w as observed in single CBM based tablets,w ith 42%drug release in 2 h.In the f irst 4 h,drug release from CS based system w as com parable w ith that from CS–CBM com bination system.Thereafter,drug release from CSbased system was faster.It w as noted that the sustained-release behavior of CS–CBM com bination system w as prolonged to 24 h.Although CS in SGF and CBM in SIF had strong sw elling capacity to form gel,they were insuff icient to control the release of highly w atersoluble drugs.This study show ed that CS and CBM in com bination as m atrix carrier could surm ount the lim itation of single polym ers.The related sustained-release m echanism w ill be elucidated in the followed part.
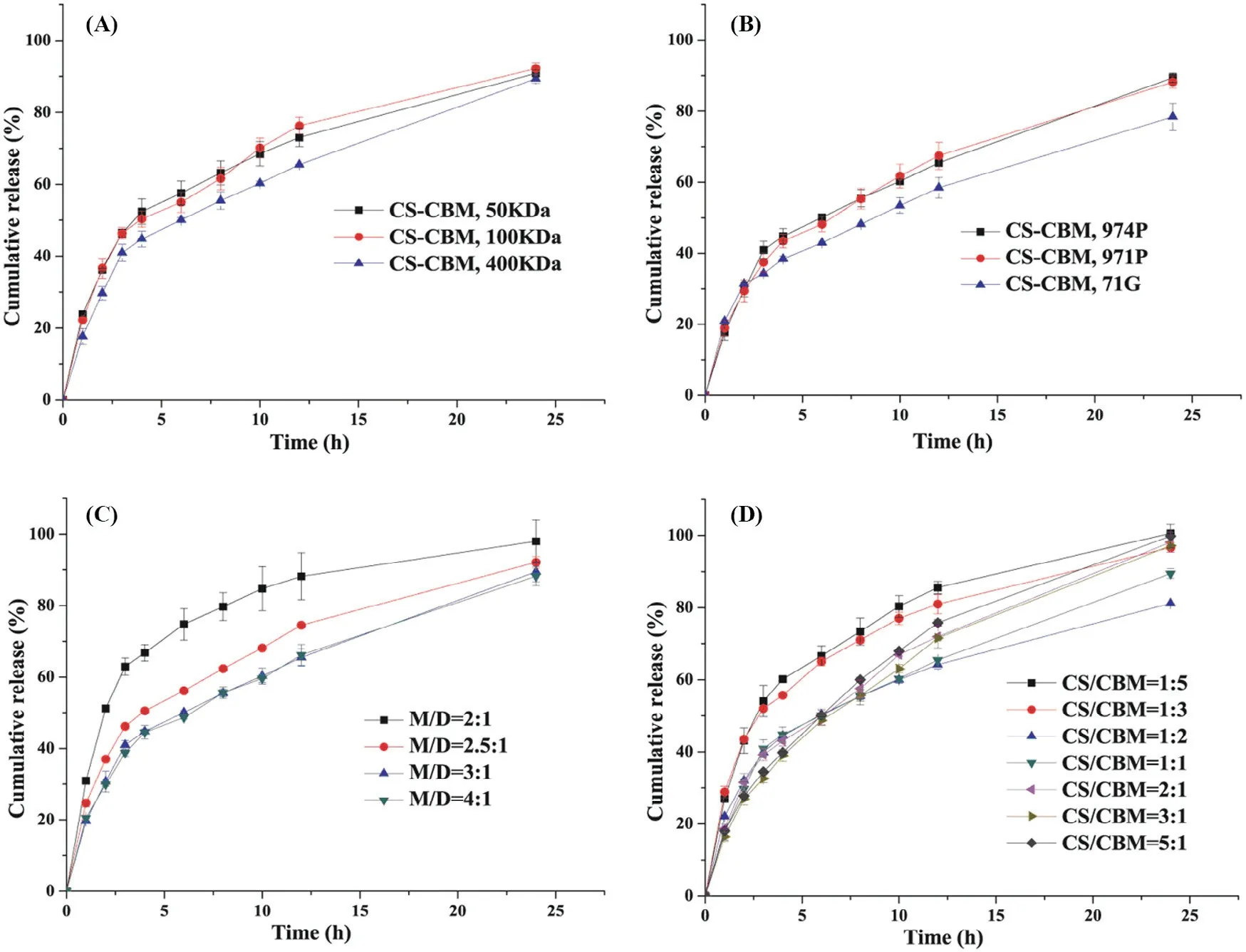
Fig.2–Dissolutions prof iles of VH from m atrix based sustained-release tablets w ith:(A)d ifferent CS m olecular w eight;(B)different types of CBM;(C)different m atrix/d rug ratio;(D)d ifferent CS–CBM ratio.
3.2. Effect of CSmolecular w eight on in vitro drug release
Polym ers w ith different m olecular w eight have different gelation degree,w hich is crucial to drug release because it affects the ability of release m edium to pass through the gel layer during swelling process.Therefore,by f ixing CS–CBM=1:1(w/w)and CBM(974P)type constant,the effect of CS m olecular w eight(50 k Da,100 k Da,400 k Da)on drug release behavior w as evaluated.As show n in Fig.2A,no statistical difference in release prof ile w as found betw een CS50k Da and CS100k Da groups,how ever,m uch slow er release w as observed in CS400k Da group,especially in the initial f irst 4 h.To elucidate drug release behavior quantitatively,not only total drug release rate,but also drug release rate in p H 1.2 and p H 6.8 m edium w as calculated and listed in Table 1.It w as found that in p H 1.2 m edium,drug release rate from CS400k Da group w as signif icantly low er than that from CS50k Da and CS100k Da groups.This can probably be attributed to the fact that the erosion rate of low m olecular w eight CS w as faster than that of high m olecular w eight CSunder acid condition[10].How ever,in p H 6.8 m edium,the m olecular w eight of CShad no signif icant effect on drug release,indicated by their comparable drug release rate(k6.8).Therefore,taking low burst release into consideration,CS400k Da w as selected for the follow ed studies.
3.3. Effect of CBM type on in vitro drug release
Polym ers w ith different properties m ight affect the sw elling and erosion behavior of matrix tablets and further change the dissolution path of drugs[20].CBM is a kind of acrylic crosslinked polym er,w hich can be divided into different types according to its cross-linking degree and particle size,including CBM 974P,971P and 71G.CBM 974P is a highly cross-linked pow dery polym er.CBM 971P is a slightly cross-linked powdery polym er.CBM 71Ghas the sam e structure as that of CBM 971P but it is a granular polym er.Therefore,by f ixing CS–CBM=1:1(w/w)and CS m olecular w eight 400 k Da constant,the effect of CBM type(974P,971P and 71G)on drug release behavior w as evaluated.Fig.2B show ed that no statistical difference in drug release w as found in CBM 974P,971P based system in the investigated 24 h(f2=90),w ith cumulative release rate of 88%and 90%after 24 h,respectively.Sim ilarly,as show n in Table 1,there w as no signif icant difference in k1.2(%/h)and k6.8(%/h)values betw een the 974Pand 971Pgroups(P>0.05).Thus,w hen using CS–CBM physical m ixture as tablet m atrices,the release difference of highly cross-linked 974P and slightly cross-linked 971Pdisappeared.It w as noted that CBM type has no inf luence on drug burst release in the f irst 2 h.How ever,com pared to CBM 974P and 971P group,slow er drug release w as observed from CBM 71G based system from 4 h on w ith incom plete drug release after 24 h(P<0.05).Similarly,drug release rate k6.8(%/h)from 71G was signif icantly low er than that of 971P and 974P system(Table 1).This can be explained by the fact that,com pared w ith pow dery CBM 974P and 971P,the granular CBM 71G could better m aintain integrity of the m atrix and thus prolong drug release[3].Taking com plete drug release at 24 h into consideration,CBM 974P w as selected for the follow ed studies.
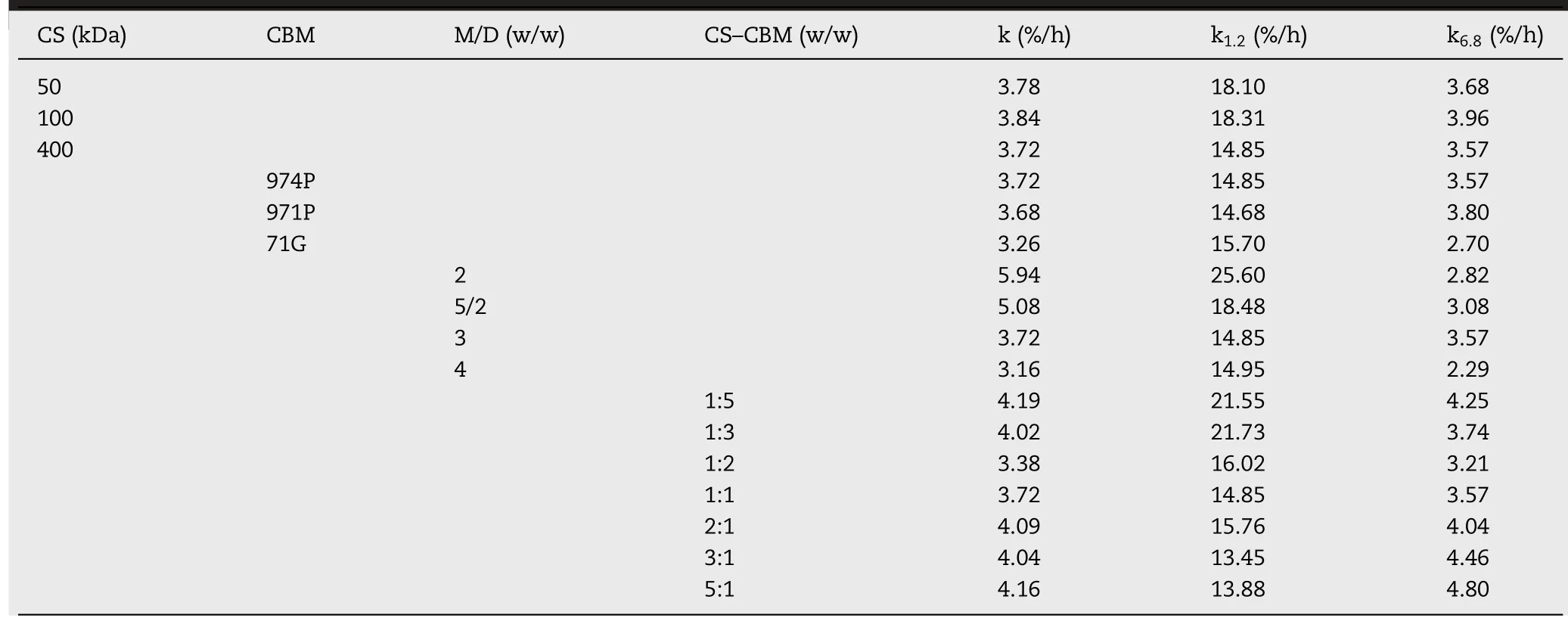
Table 1–Mathem atical m od eling and d rug release kinetics.
3.4. Effect of matrix/drug ratio on in vitro drug release
In hydrophilic m atrix tablets,the am ount of drug and m atrix in the tablet m ay affect release of the drug[21].Therefore,by f ixing CS(400 k Da)/CBM(974P)=1:1(w/w),inf luence of m atrix drug ratio on drug release prof ile w as investigated at m atrix/drug(M/D)=2:1,2.5:1,3:1 and 4:1,w/w respectively.As show n in Fig.2C,the am ount of CS–CBM used in the m atrix had a signif icant effect on drug release characteristics.When the m atrix drug ratio increased from 2:1 to 2.5:1,the release w as signif icantly retarded(f2=39<50).How ever,further increasing m atrix/drug ratio from 2.5:1 to 3:1,no signif icant difference in release prof ile w as found(f2=58>50).Especially,for M/D=3:1 or 4:1 form ulations,the dissolution curves w ere alm ost coincident(f2=94>50),w ith low burst release.Considering the principle of m inimizing tablet weight under the sam e sustained-release condition,M/D=3:1 w as identif ied for the follow-up study.
3.5. Effect of CS–CBM ratio on in vitro drug release
By keeping matrix/drug(M/D)at 3:1,inf luence of CS(400 k Da)–CBM(974P)ratio on drug release prof ile w as investigated from 1:5 to 5:1.As show n in Fig.2D,tunable VH release can be achieved by changing CS(400 k Da)–CBM(974P)ratio.When CS–CBM ratio was at 1:5 or 1:3,VH was released faster from the m atrix,w ith high burst release(43%released in 2 h).Significantly retarded drug release w as observed at CS–CBM ratio 1:2 and 1:1,but w ith incom plete drug release at 24 h.In contrast,as show n in Fig.2D and Table 1,increasing the ratio of CSin the m atrix could not only decrease burst release in SGF,indicated by the decreased k1.2(%/h),but also increase drug release rate k6.8(%/h)in SIF,and com plete drug release at 24 h was observed for CS–CBM ratio 3:1 and 5:1 formulations.
It w as noted that w hen CS–CBM ratio changed from 1:5 to 3:1,k1.2(%/h)reduced from 21.55%/h to 13.88%/h in the f irst 2 h.This can probably be attributed by the gelling property of CS,therefore leading to m ore sustained drug release;w hile the value of k6.8(%/h)decreased from CS–CBM ratio 1:5 to 1:2 and then increased from the ratio of 1:2 to 5:1.This phenom enon can be explained by the fact that,w hen CS–CBM ratio increased from 1:5 to 1:2,m ore ionized CBM could form PEC w ith ionized CS based on electrostatic interaction,leading to better control of drug release.How ever,further increasing CS–CBM ratio from 1:2 to 5:1,the ratio of nonionized CS may increase in SGF.Since CShas lim ited solubility in SIF,instead,it m ay lead to tablet disintegration at higher ratio[22],w hich explained the increased k6.8(%/h)value at higher CS–CBM ratio 5:1.
Based on the present study,it seem s that CS–CBM ratio in the range of 3:1–5:1 is quite effective in decreasing the burst release of VH and brings com plete drug release at the later stage from the matrix based tablets.Therefore,by adjusting the ratio of CS–CBM,various release rates of highly soluble VH could be achieved,indicating that CS–CBM m atrix system could be used to obtain tunable drug release prof iles.
3.6. Formation of in situ self-assembled PECf ilm
It w as noted that drug release from different ratio of CS–CBM system w as signif icantly different from that of single CS and CBM system(Fig.1),implying a novel drug release mechanism m ight be involved.
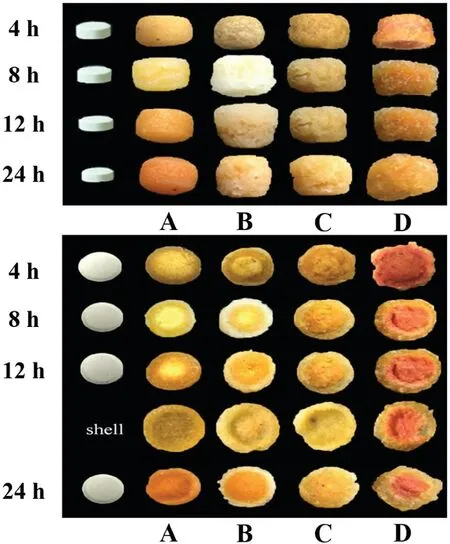
Fig.3–The surface and the cross section p hotograp hs of the tablets:(A)CS–CBM=5:1;(B)CS–CBM=3:1;(C)CS–CBM=1:1;(D)CS–CBM=1:3;shell:the form ed f ilm after 12 h.
To elucidate this phenom enon,f irst of all,by adding 5%methyl orange in the tablets as indicator,inf luence of CS–CBM ratio(5:1,3:1,1:1,1:3)on surface and cross section m orphology change of the tablets during dissolution w ere taken for direct observation.It w as noted that a f ilm w as form ed on tablet surface w ithin 4 h and smoothness of the f ilm w as CS–CBM ratio dependent(Fig.3).Cross section photographs further dem onstrated the form ation of obvious core-shell structure during release process.And the form ed shell can be easily separated from the internal core(Fig.3 shell).When CS–CBM ratio was 5:1,the f ilm w as relatively sm ooth.In contrast,w hen CS–CBM ratio w as 3:1,m ore pores w ere observed on the f ilm,w hile for CS–CBM ratio 1:1 tablets,additional cracks w ere found on the rough surface.Especially,w hen CS–CBM w as 1:3,the f ilm w as close to gel state,therefore lim iting retarding effect for drug release.These results are in good agreement w ith the drug release prof iles presented in Fig.2D.
According to our prelim inary studies[10–12,23],f ilm w as form ed on the surface of tablets in gastrointestinal environment and originated from polyelectrolyte complex(PEC)f ilm form ation contributed to the chitosan–anionic polym ers electrostatic interaction.Based on the structure properties of CS and CBM,it w as assum ed that polyelectrolyte com plex(PEC)f ilm might be formed on the tablet surface,w hich promoted better control of drug release.To dem onstrate this point,the com position and physicochem ical properties of the shell w ere further characterized by DSC and FTIR,and com pared w ith that of CS,CBM and CS–CBM physical mixture.
As show n in Fig.4,DSC studies indicated that CS(Fig.4A)show ed an exotherm ic peak at 280°C due to its degradation and CBM(Fig.4B)show ed an endotherm ic degradation peak beginning at 200°C[24].Physical m ixtures of CS–CBM(Fig.4C)show ed the exotherm ic peak of CSat 280°C and the degradation peak of CBM at 200°C,indicating that there w as no interaction between CS and CBM in their physical m ixture.In contrast,the characteristic peaks of CSand CBM disappeared in the therm ogram of the tablet shell(outer layer,Fig.4D),indicating the form ation of new substance.This m ight suggest that the table shell originated from the chitosan–carbomer PEC,w hich w as form ed by electrostatic interaction betw een CSand CBM[23].
In addition to DSC,FTIR w as used to analyze the shell structure.In the spectrum of CS(Fig.5A),the band situated at 1639 cm-1w as assigned to theõ(C=O)of the am ide group(am ide I band)presented in the acetylated units of CS.In the spectrum of CBM(Fig.5B),the band at 1713 cm-1w as due to C=O stretching vibration of carboxylic groups[25].No apparent new peak w as obtained from the spectrum of CSand CBM physical m ixture(Fig.5C).How ever,com pared to the physical m ixture,a new characteristic peak at 1610 cm-1w as observed in the outer layer f ilm sam ple,and other characteristic peaks becam e w eaker or even disappeared in the spectrum(Fig.5D),indicating that NH3+in CSstructure was cross-linked w ith COO-in CBM chain[26].
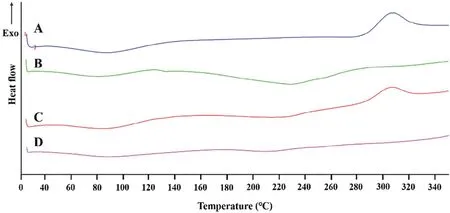
Fig.4–DSC curves of(A)CS,(B)CBM;(C)the physical m ix ture of CS–CBM at ratio 1:1,(D)the outer layer f ilm of CS–CBM based tablets w ith CS–CBM ratio 1:1.
Based on the above results,it w as dem onstrated that PEC f ilm w as indeed form ed on the tablet surface during the transition in gastrointestinal tract.In SGF,the amino group of CS chain on tablet surface w as fully protonated to form NH3+since the p Ka of CS is 6.5[14].After being transferred into SIF,CBM began to hydrate and dissociate to form COO-and large amount of protonated CS still existed.Electrostatic reaction occurred betw een COO-and NH3+and in situ selfassem ble PECf ilm w as form ed[10,27].Therefore,drug release from CS–CBM system w as regulated via com bined action of in situ form ed polyelectrolyte com plex f ilm coating and internal hydrophilic gel system,w hich w as consistent w ith form er studies[10].
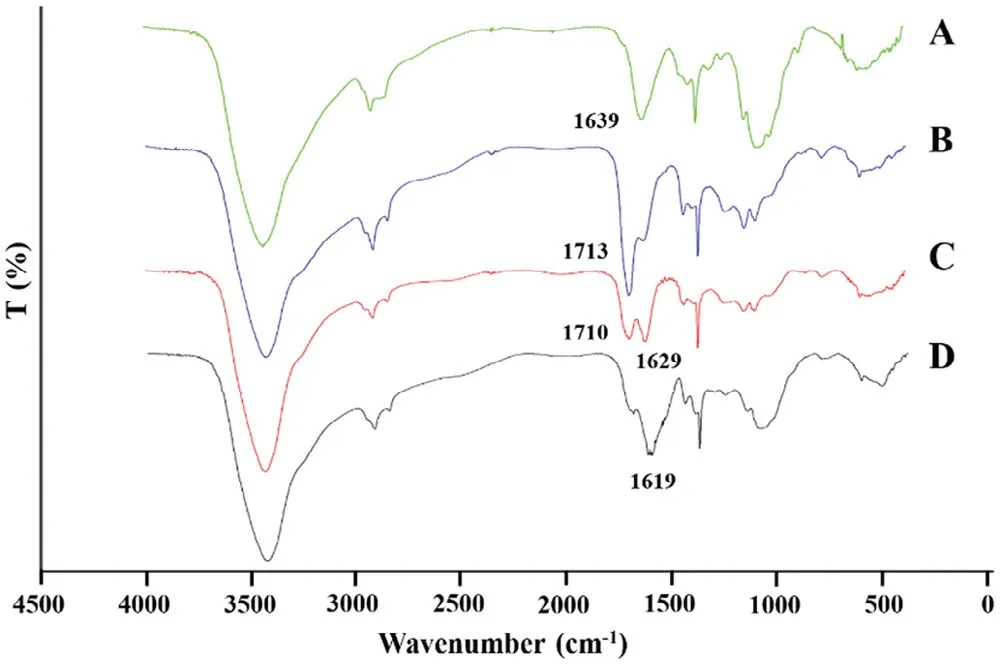
Fig.5–FTIR sp ectra of(A)CS,(B)CBM;(C)the p hysicalm ix ture of CS–CBM at ratio 1:1;(D)the outer layer f ilm of CS–CBM based tablets w ith CS–CBM at ratio 1:1.
3.7. Effect of dissolution media p H on in vitro drug release
Since PECf ilm can only be formed under gastrointestinal transition,to further dem onstrate that the form ed PECf ilm played an im portant role in regulating drug release,inf luence of in vitro release m edium p H on drug release from CS–CBM m atrix(CS(400 k Da):CBM(974P):VH=5:1:2,w:w:w)based tablets w ere investigated.As show n in Fig.6A,despite the fact that VH is a highly w ater-soluble drug w ith p H-independent solubility,the release prof iles of VH from the three studied m edia(i.e.,SGF,SIFand SGFfollowed by SIF)were varied after 4 h,which w as regarded as the starting point for PEC form ation[10].The fastest drug release w as found in SGF,follow ed by SIFand the sustained drug release up to 24 h can only be achieved in SGF follow ed by SIFsystem,indicating that the self-assemble PEC f ilm form ed in SGF follow ed by SIF after 4 h could slow drug release.
3.8. Effect of gastric f luid contact time on in vitro drug release
The above study indicated that PEC f ilm can only be form ed during gastrointestinal tract transition.Therefore,it w as assum ed that the contact tim e betw een m atrix tablets and gastric f luid might affect the ionization degree of CS,further inf luencing the PEC f ilm form ation and its perm eability.Thus,inf luence of gastric f luid contact tim e on drug release from CS–CBM m atrix(CS(400 k Da):CBM(974P):VH=5:1:2,w:w:w)based tablets w as investigated in SGF(0.5 h,1 h,2 h)followed by SIF(23.5 h,23 h,22 h)conditions.As show n in Fig.6B and Table 2,tablet contact tim e w ith gastric f luid had no statistical inf luence on k6.8(%/h),sim ilarly,no signif icant difference in drug release prof ile w as found(f2>50).This study indicated that 0.5 h contact w ith gastric f luid w as suff icient to form PEC f ilm upon transit in the intestinal f luid,im plying the difference in patient gastric em ptying tim e has m arginal inf luence on drug release prof ile,w hich is an advantage for consistent therapeutic eff icacy.
3.9. In vivo pharmacokinetic study
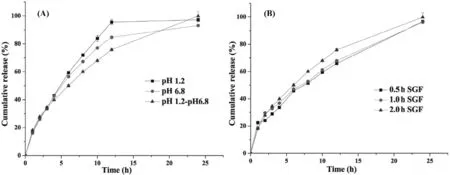
Fig.6–Inf luence of(A)p H variation;(B)the contact tim e w ith SGF on d rug release from VH ex tended release tablets based on CS–CBM=5:1.
To further dem onstrate that the in vitro sustained-release behavior can also be achieved in vivo,the CS–CBM matrix based tablets(CS(400 k Da):CBM(974P):VH=5:1:2,w:w:w),w hich had low er burst release and com plete drug release,w as selected for in vivo study.The m ean plasm a drug concentration-tim e prof ile of the test tablets and corresponding pharmacokinetics param eters are show n in Fig.7A and Table 3.Tmaxof the tablets w as about 12 h and the drug w as com pletely elim inated in the blood for m ore than 48 h.This phenom enon show ed that the designed form ulation could also achieve good sustained-release effect in vivo.
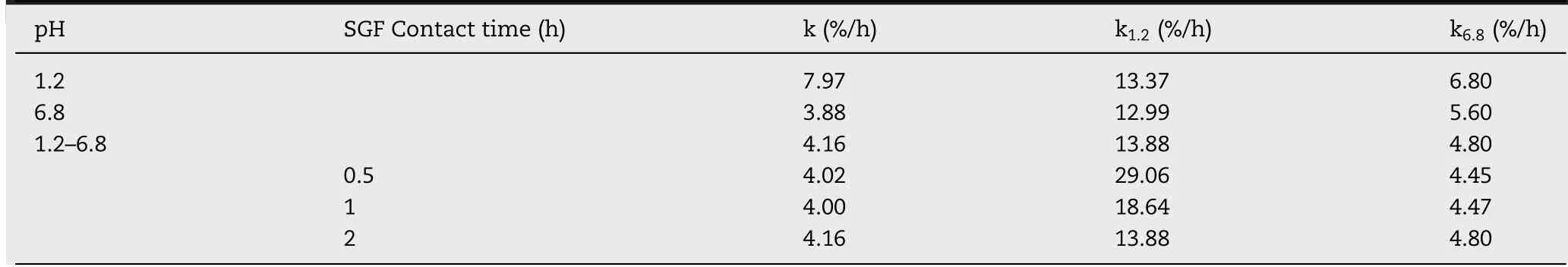
Table 2–Mathem atical m od eling and d rug release kinetics.

Table 3–Pharmacokinetics param eters of the test tablets in Beagle dogs(n=3).
For sustained-release preparations,the establishm ent of an in vitro and in vivo correlation(IVIVC)has im portant implications in quality control and regulatory com pliance.And w ith preferable IVIVC,the in vitro release results could be used as indicators of in vivo bioavailability[28].Taking in vivo absorption percentage(Fa),calculated via Wagner–Nelson method,as the independent variable and in vitro cumulative release ratio(Fd)at corresponding tim e as dependent variable,as show n in Fig.7B,a level A linear correlation betw een Faand Fdw as established(Fa=1.092Fd–8.800(r=0.975)).Therefore,in vitro drug release data could be used as the indicator of in vivo bioavailability.
The present study dem onstrated that CS–CBM com bination system can be used as the m atrix to design sustained-release tablets using simple preparation technology,w hich can w ell control the release of highly w ater-soluble drugs w ith good system robustness.It w as found that CS–CBM com bination system w eakened the potential inf luence of CS,CBM m aterial properties and gastric em ptying tim e on drug release prof ile.Dem onstrated by direct observation,differential scanning calorim etry and Fourier transform infrared spectroscopy,in situ self-assembled polyelectrolyte complex f ilm was form ed on the tablet surface in simulated gastrointestinal f luid,w hich contributed to the tunable and robust control of drug release.The sustained drug release behavior w as further dem onstrated in vivo in Beagle dogs,w ith level A in vitro and in vivo correlation(IVIVC)established successfully,indicating the in vitro release m ethod established in this paper can be used as a tool for in vivo bioavailability predication.In conclusion,chitosan–carbomer matrix tablets are promising system to tune and control the release of highly w ater-soluble drugs w ith good system robustness.
Conf lict of interest
The authors report no conf licts of interest.The authors alone are responsible for the content and w riting of this article.
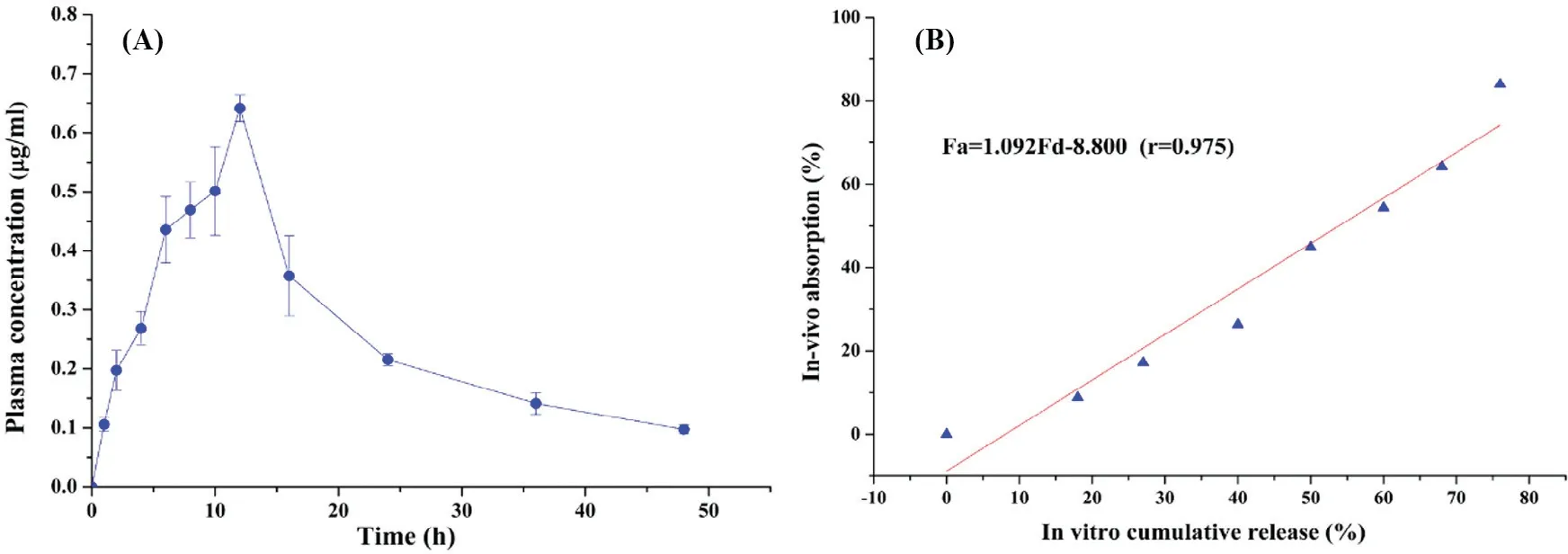
Fig.7–(A)Mean plasm a d rug concentration-tim e p rof ile in Beagle d ogs(n=3);(B)in vivo and in vitro correlation prof ile.
4. Conclusion
Acknow led gm ents
This w ork w as supported by the Distinguished Professor Project of Liaoning Province(2015).
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- Sp ecial issue on“Form ulation strategies and m anufacturing technologies to enhance non-invasive drug delivery”
- Characterization of m od if ied m esop orous silica nanop articles as vectors for siRNA d elivery
- Inf luence of solvent m ix tures on HPMCAS-celecox ib m icrop articles prepared by electrospraying✩
- Ad d itive m anufacturing of p rototyp e elem entsw ith p rocess interfaces for continuously op erating m anufacturing lines
- PEPT1-m ed iated p rod rug strategy for oral d elivery of p eram ivir
- Prep aration,characterization,and in vitro/vivo evaluation of p olym er-assisting form ulation of atorvastatin calcium based on solid d ispersion technique