磷化钴(101)面析氢性能的第一性原理研究
2018-04-18王登兴陶华龙张秋菊张志华
王登兴,陶华龙,张秋菊,张志华
(1.大连交通大学 材料科学与工程学院,辽宁 大连 116028;2.中国科学院宁波工业技术研究院 材料技术研究所,浙江 宁波 315201)
0 引言
氢气作为清洁、可再生、环境友好能源已成为近年研究热点[1-2]. 目前,最成熟的制氢方法是从化石资源通过蒸汽转化方法获得. 这不仅加剧了化石能源的消耗速度,而且蒸汽转化过程中产生的二氧化碳会进一步加剧全球的温室效应. 因此,利用可再生能源,发展环境友好的制氢方法至关重要. 通过电解水制氢是一种解决能源危机的有效途径,该反应不排放温室气体和其他污染性气体. 水的电解过程可分为两个半反应:析氢反应(HER,2H+(aq)+2e-→ H2(g))和析氧反应(OER,2H2O(l) → 4e-+4H+(aq)+O2(g)). HER性能的好坏和氢原子在电极表面吸附氢的自由能有很大关系,自由能符合火山模型,只有在自由能接近于零的时候性能最好,自由能大于零或者小于零都会使得HER的性能下降. Pt作为一种很好的电极材料,在酸性条件下有很高的析氢反应性能和反应速率,可以用来作为电极(自由能为-0.09 eV). 但是由于Pt在地球上的储量很少,Pt资源的稀缺和价格大大限制了其进一步大规模的应用. 因此找寻其他高活性、储量丰富、无毒的电极材料来替代Pt成为制氢的关键所在. 过渡金属磷化物作为新型的非贵金属,地球储量丰富,无毒、廉价,可以用来替换Pt电极材料. 根据实验和理论研究[3-5],磷化钴有很好的析氢反应性能. 本文计算了不同3d金属元素掺杂CoP(101)面的自由能,研究了不同掺杂位点和掺杂浓度对其自由能的影响,对掺杂的磷化钴作用机理进行了电荷和态密度的理论分析.
1 计算方法
所有的密度泛函理论(DFT)计算通过VASP(Vienna ab initio simulation package)软件来实现[6-7]. 对于电子之间的交换函数,使用广义梯度近似GGA(generalized gradient approximation)中的PBE(Perdew-Burke-Ernzerhof)方法[8],原子和原子核之间的相互作用采用PAW(projector-augmented-wave)方法来实现[9-10],平面波的截断能设置为450 eV. 当原子间每个离子的受力小于0.02 eV/Å,总的能量变化小于10-4eV时弛豫结束. K点网格选取基于Monkhorst-Pack方法[11]. 为了尽可能减小表面模型之间的相互作用,沿着c轴的真空层取15 Å.
HER活性和电催化剂表面吸附氢的自由能密切联系[12-13],在催化剂表面吸附氢的自由能可以根据以下公式计算:
ΔGH*=Etotal-Esur-EH2/2 + ΔEZPE-TΔS

2 实验结果与讨论
2.1 CoP(101)面的析氢性能研究
CoP块体是B31或者MnP-类型的结构,空间群为Pnam. 在这种结构中,每一个金属钴原子被六个磷原子围在一个扭曲的八面体结构中心,如图1(a)所示. 而每一个磷原子周围是六个金属钴原子,形成一个高度扭曲的三角棱形(图1(b)).计算的晶格参数a=5.053 Å,b=3.269 Å,c=5.540Å,与实验晶格参数[14]a=5.077 Å,b=3.281 Å,c=5.587 Å基本一致. Co-P键长范围为2.199 Å-2.348 Å,平均键长大约为2.292 Å.
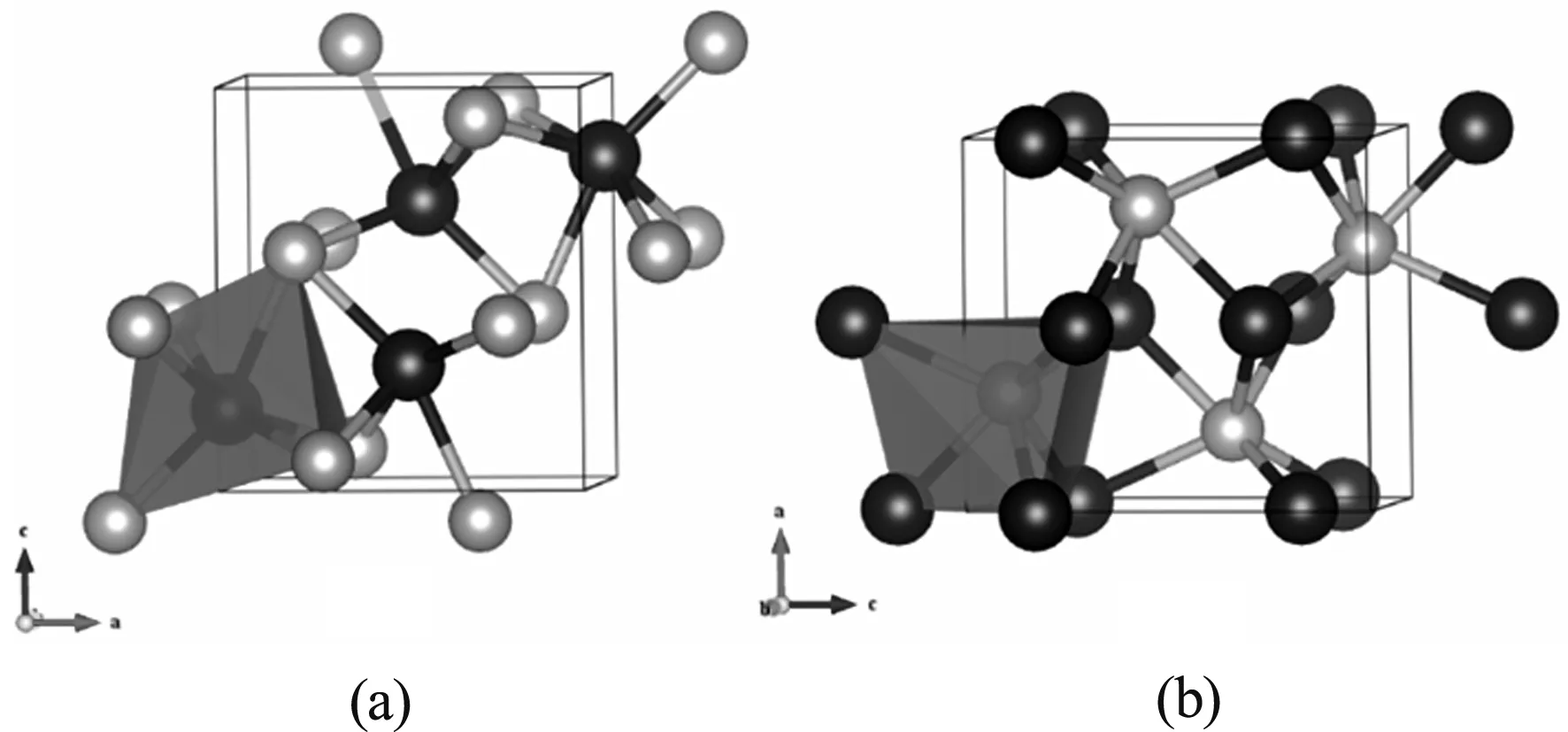
图1 CoP块体结构
根据块体模型的结构,参考已有研究结果[3-4, 15],本文选取CoP(101)面来研究氢原子在其表面的吸附. CoP(101)面的模型如图2(a,b)所示,(a)和(b)分别为侧视图和表面两层俯视图. 表面有两种钴环境,一种Co是在四元环中称为A,一种是在六元环中称为B,第二层的钴环境称为C. 计算时取2×1的超胞并取k点3×3×1. 该模型有六层,每一层都是4个磷4个钴. 以表面为例,其中钴有两种环境,一种钴在四元环中称为A(A-P-A-P),Co-P键长为2.344 Å和2.216 Å, 且两个∠P-A-P键角均为87.97°,
∠A-P-A键角分别为88.37°和94.97°. 一种钴在六元环中称为B(A-P-B-P-A-P-B-P),Co-P的键长分别为2.210 Å和2.233 Å,第二层的钴对表面氢吸附能影响很小,因此在第二层的钴统称为C,如图2(a),2(b)所示. 在CoP (101)面不同位置吸附氢原子,其中Co顶位的吸附能为0.247 eV,P顶位的吸附能为-0.042 eV,Co-P桥位的吸附能为0.252 eV,Co-Co(B位置)桥位的吸附能为-0.380 eV,而Co-Co(A位置)吸附位点不稳定,这表明在两个B位置桥位的吸附最稳定,且自由能为-0.14 eV. 最稳定的吸附如图2(c)所示,氢和表面吸附两个钴的键长为1.726 Å,而且键角∠Co-H-Co和∠H-Co-Co分别为112.82°和33.60°. 这些结果表明CoP有很好的HER性能,为了进一步提高CoP的析氢反应性能,考虑通过掺杂3d金属元素来改性CoP的析氢反应性能.

图2 CoP(101)面结构
由于Co元素处在元素周期表3d金属周期内,周期内的大多数元素半径接近,掺杂同周期的3d金属元素有可能改变CoP的HER性质,而且这些元素通过掺杂改性也有可能在实验上真正实现. 这里我们通过掺杂Cr、Fe、Ni、Cu、Ga等和Co半径接近的元素来改性. 替换一个钴原子的浓度为4.2%,考虑(A,B,C)三种情况;替换2个钴原子时掺杂浓度为8.3%,考虑(AA, AB, BB, AC, BC, CC)6个不同位置的掺杂情况.
掺杂这些元素后计算出的自由能在表1列出.
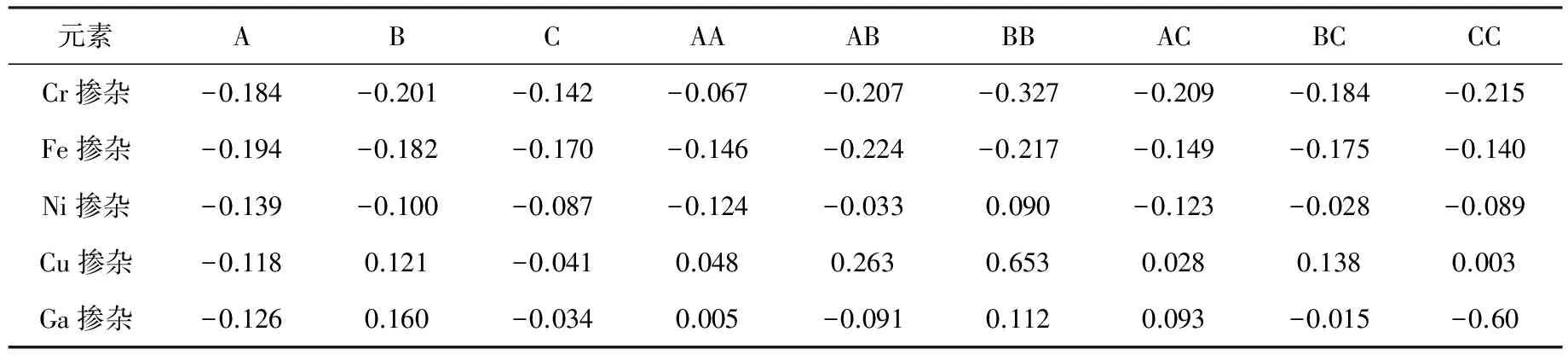
表1 掺杂Cr,Fe,Ni,Cu,Ga元素以及掺杂不同位置和不同浓度后的自由能 eV
2.2 Ni掺杂CoP(101)面的析氢性能
对于4.2% Ni掺杂CoP(101)来说,掺杂A位置、B位置和C位置都会稍微提高自由能,其中在C位置的自由能最接近零(-0.087 eV). 表明掺杂在第二层比第一层CoP(101)面有更好的HER性能. 进一步增加掺杂浓度,对于8.3% Ni掺杂来说,其中在BC位置掺杂自由能最接近零(-0.028eV),结构如图3(a)所示. Co-P键长为1.670 Å,Ni-P键长为1.764 Å,∠Ni-H-Co为114.30°. 根据Bader电荷分析(表2),吸附位点的Co和Ni分别失去0.348和0.233个电子, Co相对于没掺杂时失去的0.292电子相比,又失去了0.056个电子,Ni相比原来的Co多了0.059个电子,导致吸附氢这一区域电子增多,对氢的吸附能明显减弱. 不同浓度掺杂的自由能在图3(b)中示出. 掺杂后的态密度图在图3(c)画出.
表2纯CoP(101)以及Ni、Cu、Ga最优掺杂位置的吸附位点的Co失电子数
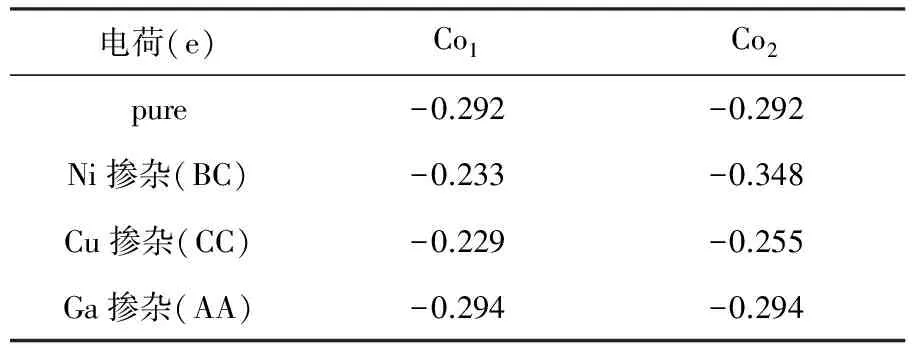
电荷(e)Co1Co2pure-0.292-0.292Ni掺杂(BC)-0.233-0.348Cu掺杂(CC)-0.229-0.255Ga掺杂(AA)-0.294-0.294
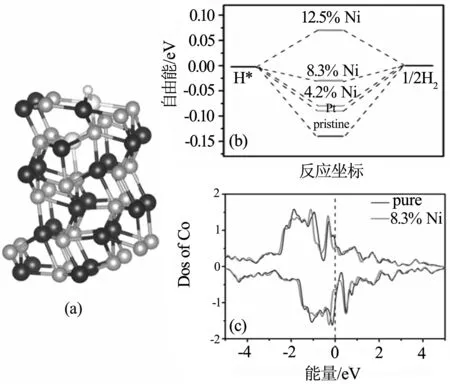
图3 8.3% Ni掺杂CoP(101)(BC位置)
可以看出随着掺杂浓度的增加,自由能随着增加. 自由能进一步增加越过火山模型的顶点,使得HER的性能变坏. 说明Ni掺杂浓度在8.3%左右有最好的析氢性能(自由能为-0.028 eV).
2.3 Cu掺杂CoP(101)面的析氢性能研究
对于4.2% Cu掺杂CoP(101)来说,A, B, C三个位置中最优位置和Ni一样. C位置自由能最接近零且自由能为(-0.041 eV). 对于8.3% Cu 掺杂CoP(101)来说,CC位置的自由能最接近零(0.003 eV),如图4(a)所示. H和表面两个Co的键长分别为1.715和1.704 Å,∠Co-H-Co为112.47°. 根据Bader电荷分析(表2),吸附位置的2个Co分别失去0.229和0.255电子,相对于CoP没掺杂前的Co失去0.292个电子,说明掺杂Cu后2个Co平均获得0.051个电子,减弱了氢的吸附. 图4(b)也说明随着掺杂Cu浓度的提高,CoP的自由能会先提高后降低,且掺杂8.3%的Cu的CoP有最高的析氢反应性能. 而且从最优位置(CC)的态密度(图4(c)), 说明掺杂Cu后表面吸附氢能力减弱,从而提高其HER性能.

图4 8.3% Cu掺杂CoP(101) (CC位置)
2.4 Ga掺杂CoP(101)面的析氢性能
另外我们也进行了Ga掺杂CoP(101)面的析氢性能研究. 对于4.2% Ga掺杂的CoP,C位置同样是性能最好的掺杂位点且自由能为(-0.034 eV). 而8.3%掺杂的浓度,六个位置掺杂性能最好的是AA位,P和表面两个Co的键长分别为1.732和1.736 Å,键角∠Co-H-Co为113.91°,自由能为0.005 eV. 根据Bader电荷分析,此时两个Co失去0.294个电子,相对于没掺杂前的Co失去0.292电子,Co又失去0.002电子,但是由于表面有Ga的存在,而Ga的最外层电子比Co多,因此掺杂后表面还是多电子,所以吸附能会降低. 图5(c)列出了掺杂后的表面Co原子的态密度. 自由能随掺杂浓度的变化和Ni、Cu一样符合火山模型,掺杂浓度为8.3%时,自由能最接近零,浓度太低或者太高都会降低其析氢反应的性能.
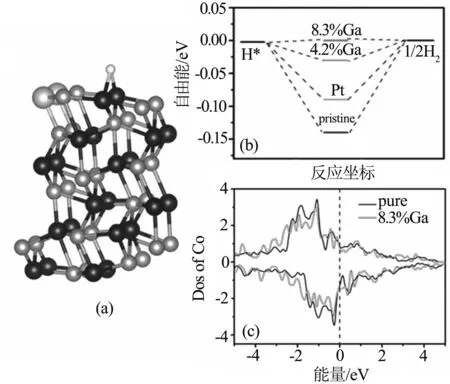
图5 8.3%Ga掺杂CoP(101) (AA位置)
2.5 Cr、Fe掺杂CoP(101)面的析氢性能
根据表1的计算结果,发现掺杂Cr、Fe(元素周期表Co左边的元素)会使得CoP的HER性能下降. 这可能是由于Co左边的Cr、Fe元素比Co少电子,掺杂这些元素后造成体系的电子减少,增强了对氢的吸附能力. 而掺杂Ni、Cu、Ga(Co右边的元素)会使得CoP的HER性能有所提高,这由于Co右边的Ni、Cu、Ga元素最外层比Co多电子,掺杂这些元素后使得体系的电子增多,减弱了对氢的吸附能力,使得HER的性能有所提高. 对不同的元素掺杂量和不同的掺杂位置进行分析,结果表明随着Ni、Cu、Ga的浓度提高,HER性能会先提高后降低,因此掺杂的浓度不能超过10%.
3 结论
为了阐明CoP的析氢反应性能,本文计算了CoP(101)面以及掺杂(Cr, Fe, Ni, Cu, Ga)CoP(101)面吸附氢的自由能,结果表明掺杂Cr,Fe后CoP(101)面的自由能会下降,而掺杂Ni、Cu、Ga会提高CoP(101)面的自由能,但掺杂Ni、Cu、Ga的浓度不能超过10%. 掺杂不同位置和不同浓度对CoP(101)面的自由能影响也不同,掺杂8.3% Ni、Cu、Ga会使得CoP的自由能最接近零,析氢反应性能最优. 这些结果为研究CoP和其他磷化物的析氢性能提供了理论参考.
参考文献:
[1]LU Y. Review and Prospect of Clean, Renewable Energy Utilization[J]. Sci. & Tech. Rev., 2014 (32):15-26.
[2]吴新平. 我国新能源发展及应用探讨[J]. 电气时代,2015 (2): 33-36.
[3]HA D H. Activity and stability of cobalt phosphides for hydrogen evolution upon water splitting[J]. Nano Energy, 2016 (29):37- 45.
[4]HU G, TANG Q, JIANG D E. CoP for hydrogen evolution: implications from hydrogen adsorption[J]. Phys. Chem. Chem. Phys., 2016 (18): 23864-23871.
[5]KIBSGAARD J. Designing an improved transition metal phosphide catalyst for hydrogen evolution using experimental and theoretical trends[J]. Energy & Environ. Sci., 2015 (8) :3022-3029.
[6]KRESSE G, HAFNER J. Ab-initio molecular-dynamics simulation of the liquid-metal amorphous-semiconductor transition in germanium[J]. Phys. Rev. B., 1994 (49): 14251-14269.
[7]KRESSE G. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J]. Phys. Rev. B., 1996 (54): 11169-11186.
[8]PERDEW J P. Atoms, molecules, solids, and surfaces-applications of the generalized gradient approximation for exchange and correlation[J]. Phys. Rev. B., 1992 (46): 6671- 6687.
[9]BLOCHL P E. Projector augmentel-wave method[J]. Phys. Rev. B., 1994 (50): 17953-17979.
[10]KRESSE G, JOUBERT D. From ultrasoft pseudopotentials to the projector augmented-wave method[J]. Phys. Rev. B., 1999 (59):1758-1775.
[11]MONKHORST H J, PACK J D. Special points for brillouin-zone interrations[J]. Phys. Rev. B., 1976 (13):5188-5192.
[12]ZHENG Y. Advancing the Electrochemistry of the Hydrogen-Evolution Reaction through Combining Experiment and Theory[J]. Angew. Chemie-Inter., 2015(54): 52- 65.
[13]CABAN ACEVEDO M. Efficient hydrogen evolution catalysis using ternary pyrite-type cobalt phosphosulphide[J]. Nature Mater, 2015 (14):1245-1251.
[14]RUNDQVIST S. Phosphides of B31 (MNP) structure tpe[J]. Acta Chem. Scand, 1962 (16):287.
[15]SHI Y. ZHANG B. Recent advances in transition metal phosphide nanomaterials: synthesis and applications in hydrogen evolution reaction[J]. Chem. Soc. Rev., 2016 (45): 1529-1541.
