漂浮固化分散液液微萃取/液相色谱法测定人体尿液中的6种羟基多环芳烃
2018-04-18崔君涛郭溪香曾祥英于志强
李 佩,崔君涛,郭溪香,曾祥英,于志强*
(1.中国科学院广州地球化学研究所 有机地球化学国家重点实验室 广东省环境资源利用与保护重点实验室,广东 广州 510640;2.中国科学院大学,北京 100049)
多环芳烃(PAHs)广泛存在于环境中,主要来源于化石燃料、生物质等有机物质的不完全燃烧[1]。PAHs具有致癌、致畸和致突变效应,对人体健康和生态系统具有危害作用[2-3]。20世纪70年代,美国环保署(USEPA)将16种PAHs列为优先控制污染物。我国也将7种PAHs列入“优先污染物黑名单”。近年来,人体暴露于多环芳烃已受到了广泛关注[3-6]。
人们可通过呼吸空气、摄入灰尘和食物、皮肤吸收等多种途径暴露于PAHs[4,7]。进入人体的PAHs在P450酶的作用下,通过Ⅰ相代谢反应生成羟基化代谢物,也可继续通过Ⅱ相代谢反应与葡萄糖苷酸或/和硫酸酯结合形成水溶性更强的结合态代谢物,继而通过人体尿液或粪便排出体外[8]。人体尿液中的羟基多环芳烃(OH-PAHs)被广泛用作人体暴露于PAHs的生物标志物,其中1-羟基芘(芘的代谢物)是最常用的生物标志物。但研究表明,同时检测尿液中多种羟基多环芳烃对于全面评估人体暴露于PAHs非常有必要[9-10]。随着流行病学的发展,大样本量尿液的检测需求也对尿液中代谢物的检测方法提出新的要求。因此,建立同时检测尿液中的多种OH-PAHs的简单快速的分析方法具有重要意义。
目前,分析尿液中OH-PAHs的前处理方法有固相萃取[11-12]、在线固相萃取[13]、液液萃取[14]、搅拌棒吸附萃取[15]等,它们常与液相色谱/荧光检测器(LC/FLD)、液相色谱-串联质谱(LC-MS/MS)等检测技术联合分析尿液中的多种OH-PAHs。尽管这些方法均能基本满足检测需求,但对于大样本量尿液的检测有一定的局限性。如固相萃取的有机溶剂用量大,在线固相萃取和搅拌棒吸附萃取需特定的实验装置,成本较高等。2006年,Assadi等[16]开发了液液分散微萃取的方法提取水溶液中的痕量有机物,该方法具有操作简单快速等优点。但由于需收集萃取管底部的萃取剂,当样品基质较复杂时,收集过程带来的杂质干扰较多。因此,Yamini等[17]提出漂浮固化液液微萃取技术(DLLME-SFO),该方法使用低毒性的萃取剂,在低温下凝固后漂浮在萃取管表层,易于收集。目前,此方法已成功用于水[18]、化妆品[19]和尿液[20]等液体样品基质中痕量有机物的测定。本文首次使用DLLME-SFO方法萃取尿液中6种OH-PAHs,并结合液相色谱/荧光检测器(LC/FLD)技术,测定了人体尿液中的2-羟基萘(2-NAP)、2-羟基芴(2-FLU)、2-羟基菲(2-PHE)、3-羟基菲(3-PHE)、4-羟基菲(4-PHE)和1-羟基芘(1-PYR)。
1 实验部分
1.1 试剂与仪器
6种OH-PAHs标样:2-NAP、2-FLU、2-PHE、3-PHE、4-PHE和1-PYR(50 mg/L,溶于甲苯)均购自美国Supelco公司;β-葡萄糖苷酸-芳基硫酸酯混合酶(124400β-glucuronidase unit/mL,36010 sulfatase unit/mL)(美国Aldrich-Sigma公司);氯化钠、醋酸钠和冰醋酸均为色谱纯(99.97%,美国Tedia公司);盐酸(质量分数37%,德国Sigma-Aldrich公司);正十一醇、甲醇和乙腈均为色谱纯(99.9%,德国Merck公司);丙酮(农残级,99.0%,美国Honeywell公司);实验用水为超纯水(18.2 MΩ·cm),由锐思捷科学仪器有限公司的实验室超纯水系统制备。
安捷伦1200型高效液相色谱配备荧光-二极阵列管检测器(HPLC/FLD,美国Agilent公司);液相分析柱(Eclipse Plus-C18250 mm×4.6 mm i.d.,5 μm;美国Agilent公司);离心机(上海安亭科学仪器厂)。
1.2 实验方法
1.2.1标准储备液配制将OH-PAHs单标均溶于甲苯,质量浓度均为50 mg/L,作为储备液;使用微量取样针取不同体积的OH-PAHs各单标储备液,用甲醇逐级稀释至所需浓度。
1.2.2尿液的酶解尿液从冷藏室取出后放至室温,3 000 r/min离心后,准确移取上层清液5 mL,于10 mL具塞玻璃离心管中,加0.5 mL 1.0 mol/L的盐酸和1.5 mL 0.5 mol/L醋酸钠和醋酸的缓冲溶液,调节尿液的pH值为5.0;然后加20 μLβ-葡萄糖苷酸-芳基硫酸酯酶,混匀后置于恒温振荡箱中避光振荡16 h,恒温37 ℃,振荡速度100 r/min。尿液的酶解使尿液中呈葡萄糖苷酸/硫酸酯结合态的代谢物水解形成羟基代谢物。
1.2.3漂浮固化分散液液微萃取酶解后的尿液加入20%的氯化钠(质量分数)调节离子浓度。取混合均匀的萃取剂(40 μL 正十一醇)和分散剂(400 μL 丙酮),快速注入已处理好的尿样中,使之迅速形成三相浑浊的溶液,涡旋混匀后,离心5 min(3 000 r/min),此时,密度较轻的正十一醇形成一微小液滴悬浮于液体表面,然后将样品置于-20 ℃冰箱中冷冻约10 min,待正十一醇固化后,将其取出至进样细胞瓶中,室温下溶解后进液相色谱检测。
1.2.4仪器分析液相色谱分析柱采用Agilent Zorbax Eclipse Plus C18柱(250 mm× 4.6 mm,5 μm)。采用甲醇(A)-水(B)梯度洗脱:0 ~5 min,60%A;5 ~14 min,60%~78%A;14 ~21 min,78%~85%A;21 ~30 min,85%~100%A;30 ~35 min,100%A;35 ~39 min,100%~60%A;39 ~45 min,60%A。流速为0.6 mL/min,柱温为25 ℃,进样量为5 μL。各目标化合物的最佳荧光条件分别为(激发波长/发射波长):2-NAP (227/355 nm)、2-FLU (272/336 nm)、2-PHE (254/369 nm)、3-PHE (250/358 nm)、4-PHE (246/371 nm)、1-PYR (239/392 nm)。
2 结果与讨论
2.1 萃取条件的优化
2.1.1萃取剂的影响该漂浮固化分散液液微萃取的萃取剂需满足以下条件:同时溶于分散剂和尿液,密度比水小,凝固点较高,熔点较低。文献中常用高醇(如壬醇、癸醇、十一醇和正十二醇)作为萃取剂。正十一醇具有较低的水溶性,萃取过程中稳定且具有很好的萃取效果[17-18,21]。因此本研究采用正十一醇作为萃取剂。
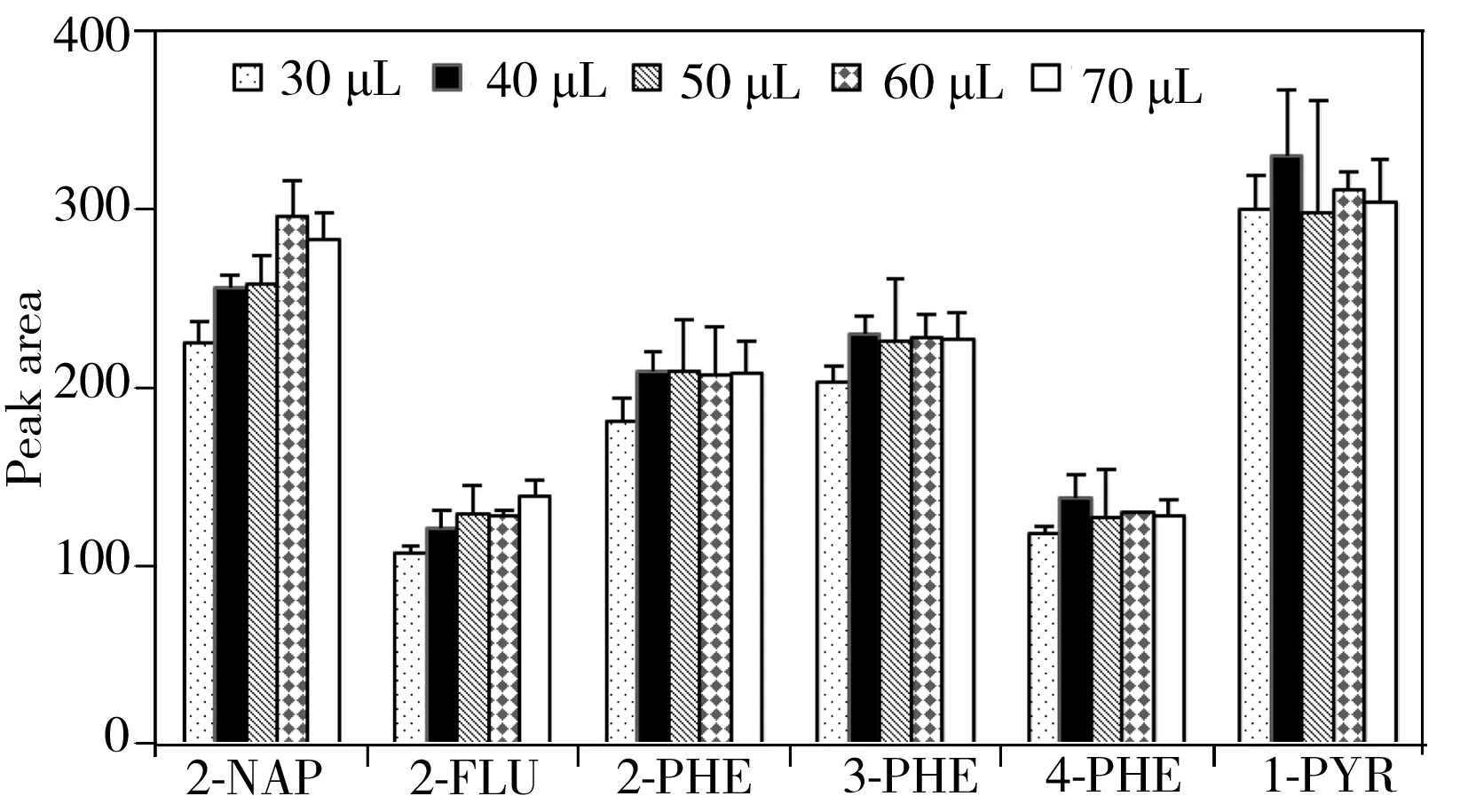
图1 萃取剂体积对萃取效率的影响Fig.1 Effect of the volume of extraction solvent (1-undecanol) on peak areas of OH-PAHs
萃取剂的体积是影响化合物萃取效率和富集倍数的关键因素。实验考察了正十一醇的体积分别为30、40、50、60、70 μL时对OH-PAHs萃取效率的影响,结果如图1所示。实验过程中发现当正十一醇的体积在30 μL及以下时,固化的上浮溶剂体积太小,很难被收集。当体积增加至40 μL时,各化合物的萃取效率明显提升,当继续增加萃取剂的体积时,对PHE和PYR代谢物的萃取效果影响较小,而2-NAP和2-FLU的回收率有所增加,这可能由化合物的极性所致,2-NAP和2-FLU的极性大于PHE和PYR的代谢物,因此更易溶于正十一醇。另外,萃取剂的使用体积增大,目标物的富集倍数减小,从尿液中带来的基质干扰也增大。因此选择40 μL正十一醇为萃取剂。
2.1.2分散剂的影响分散剂须满足以下条件:能与尿液和萃取剂形成三相互溶的乳浊状液体,增加化合物和萃取剂的接触面积,以利于尿液中OH-PAHs更好地溶于萃取剂中。实验考察了甲醇、乙腈和丙酮3种溶剂作为分散剂时对OH-PAHs萃取效率的影响(图2),发现丙酮作为分散剂的效果最好,这可能是由于丙酮的混溶性比其它两种溶剂更好。
不同体积的分散剂影响三相乳浊状液体的分散程度,分散剂太少不利于乳浊状的形成,分散剂过多不利于OH-PAHs溶于萃取剂,因此实验考察了分散剂的体积分别为200、300、400、500、600 μL时对目标物萃取效率的影响,结果显示,当分散剂体积为400 μL时,各目标物的萃取效果最好。
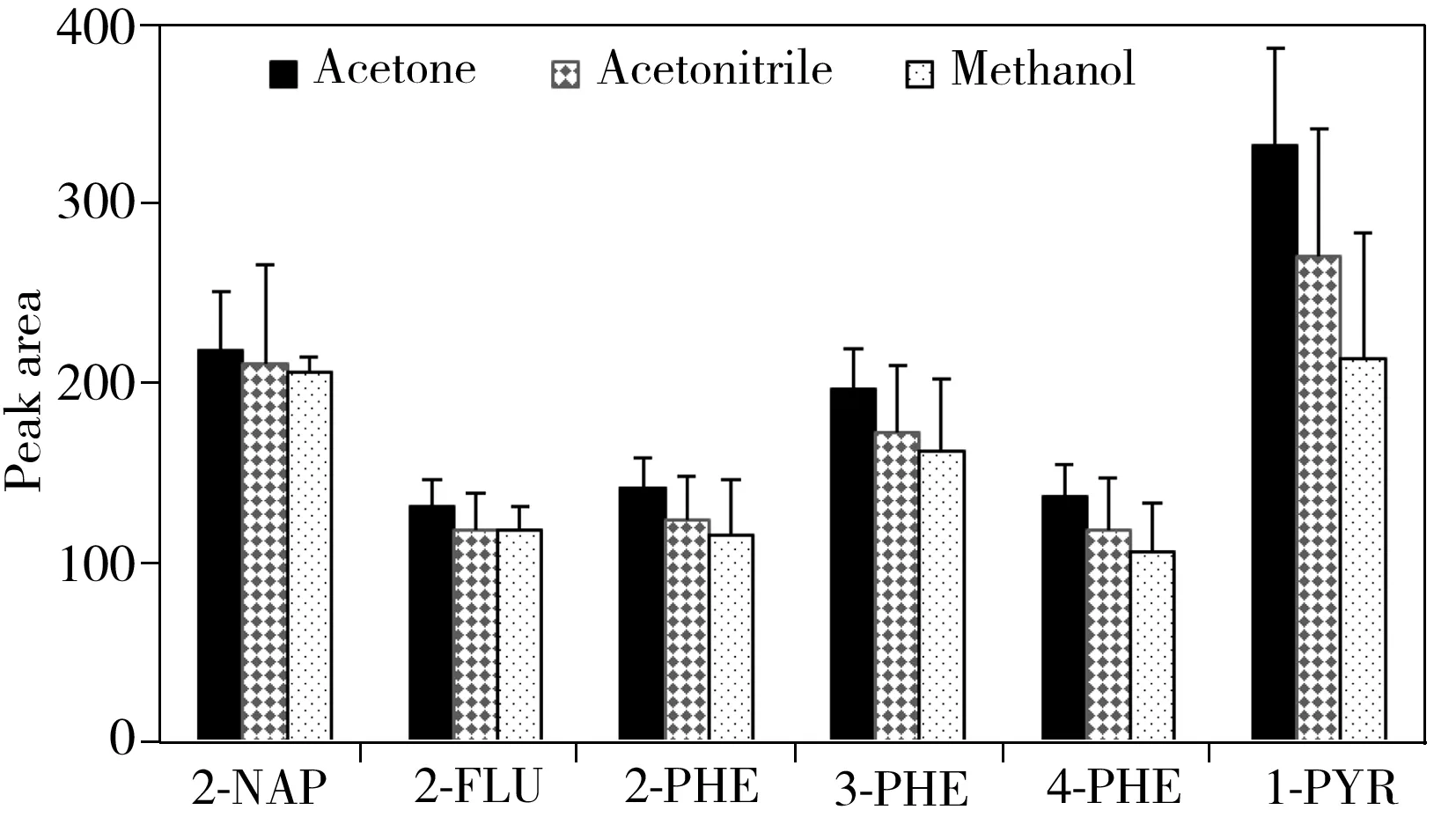
图2 分散剂种类对萃取效率的影响Fig.2 Effect of the type of dispersive solvent on peak areas of OH-PAHs
2.1.3尿液pH值的影响通过调节尿液的pH值,增加或减少尿液中质子的总量,优化OH-PAHs在溶液中以分子或离子状态存在的比例,可能有利于OH-PAHs的萃取。通过添加0.1 mol/L盐酸或氢氧化钠调节尿液的pH值范围为2.0 ~9.0,考察了pH值对萃取率的影响,结果如图3所示。在pH 5.0时,即酶解后不调节尿液的pH值,萃取效果最佳。这与文献中报道的用搅拌吸附萃取OH-PAHs的条件一致[15]。
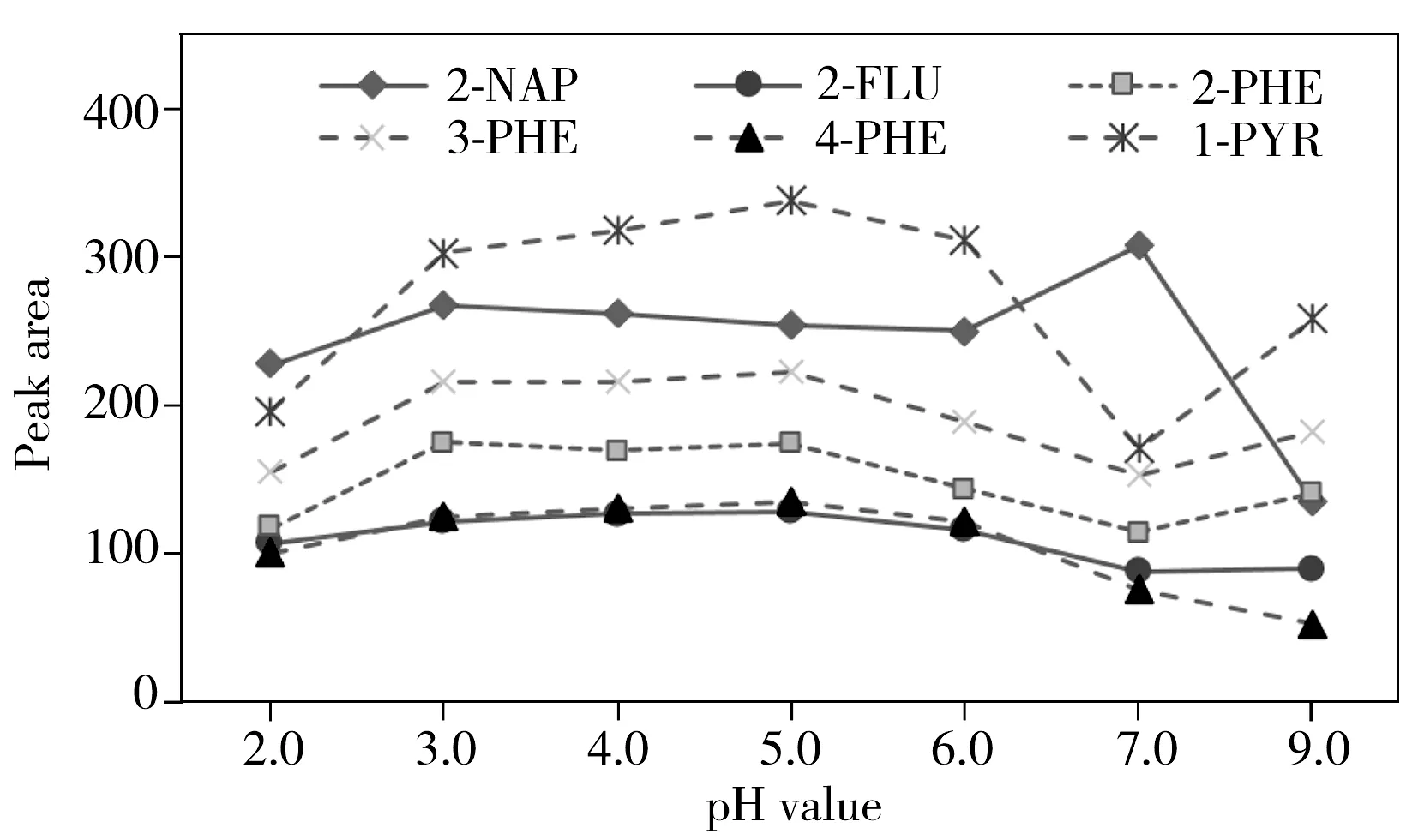
图3 尿液pH值对萃取效率的影响Fig.3 Effect of the pH value on peak areas of OH-PAHs
2.1.4盐浓度的影响在尿液中添加盐溶液增加尿液中的离子强度,有利于OH-PAHs以分子形式存在于尿液中,能更好地溶于萃取剂。实验考察了分别添加质量分数为5%、10%、15%、20%、25%的NaCl对目标物萃取效率的影响,结果显示,当添加20%的NaCl时,各化合物的萃取效果最好。
2.1.5萃取时间的影响在注入分散剂和萃取剂后,尿液迅速形成浑浊的液体,实验比较了萃取时间分别为0 (不静置,直接离心)、2、5、10 min时对目标物萃取效率的影响。结果显示,萃取时间对化合物的萃取效率无影响。表明液液分散萃取过程非常迅速,在分散剂和萃取剂注入形成浑浊液体后,目标物即被萃取出来。
2.2 方法评价
方法评价中所用的尿液基质为采集的10例志愿者尿液等体积混合后的尿液。本方法的回收率采用标准加入法进行考察,由于混合尿液基质中广泛存在OH-PAHs,因此同时进行加标基质和空白基质的前处理,每个加标浓度进行6次重复实验。结果显示,所有目标化合物在10、50、250 μg/L加标浓度下的回收率为70.4%~129%,相对标准偏差(RSD)为1.7%~14.8%,方法的准确度和精密度可满足尿液中OH-PAHs的分析要求(表1)。其中2-NAP在10 μg/L加标水平时,检测的波动稍大,这可能由于低浓度下的基质干扰较大。分别以3倍信噪比计算得检出限(LOD)为0.5 ~1.0 μg/L,以10倍信噪比计算得定量下限(LOQ)为1.5 ~3.0 μg/L,所有目标化合物在2.5~150.0 μg/L范围内均呈良好的线性关系,相关系数(r2)为0.999 1~0.999 8(表2)。方法的灵敏度可满足实际样品的检测需求。
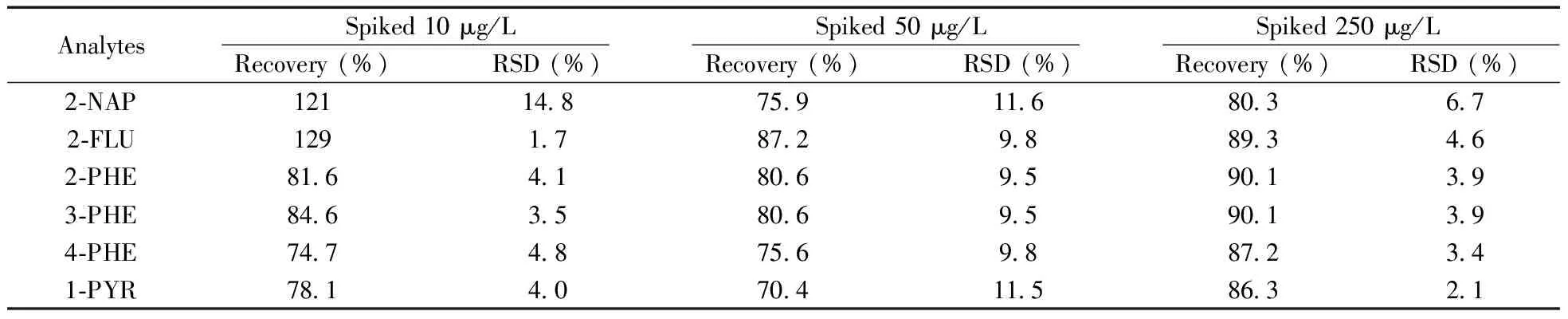
表1 不同加标浓度下各OH-PAHs的回收率和相对标准偏差(n=6)Table 1 Recoveries and RSDs of OH-PAHs in human urine spiked at different concentrations(n=6)
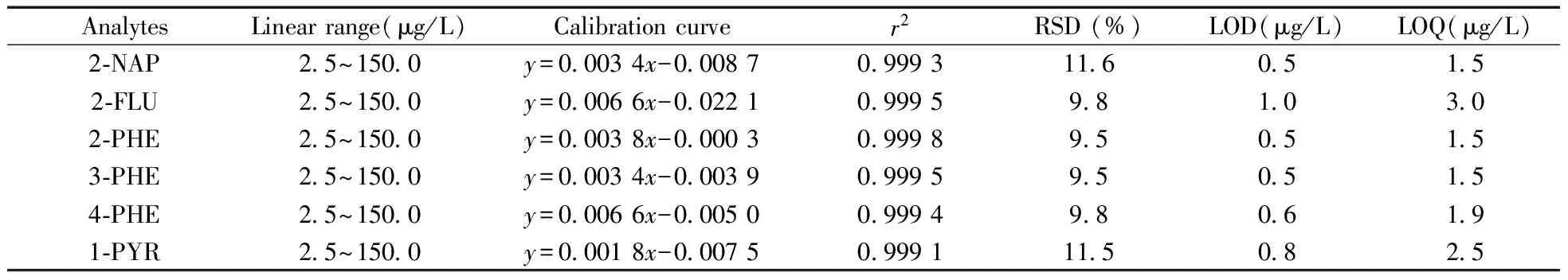
表2 DLLME-SFO方法检测尿液中OH-PAHs的线性关系、重复性、检出限与定量下限Table 2 Analytical performance data for determination of OH-PAHs in urine by DLLME-SFO method
xrepresents the response area,yrepresents the quality of analyte
2.3 与其他方法的比较
该方法与文献报道方法的比较见表3,结果表明,与传统的固相萃取[23-24]和自动液液萃取[14]相比,该方法无需特殊的实验装置,前处理时间短。虽然本方法的检出限高于使用质谱检测方法的检出限[14,23],但荧光检测器对于大多数实验室来说,价廉易得,有利于方法的推广使用。该方式使用的萃取剂正十一醇仅需几十微升,且几乎无毒,对环境友好,减少了对实验人员的健康危害。总之,相比于文献方法,该方法简单快速,灵敏度高,可满足尿液中OH-PAHs的检测需求;易于推广,对大样本量尿液中的OH-PAHs检测具有应用前景。
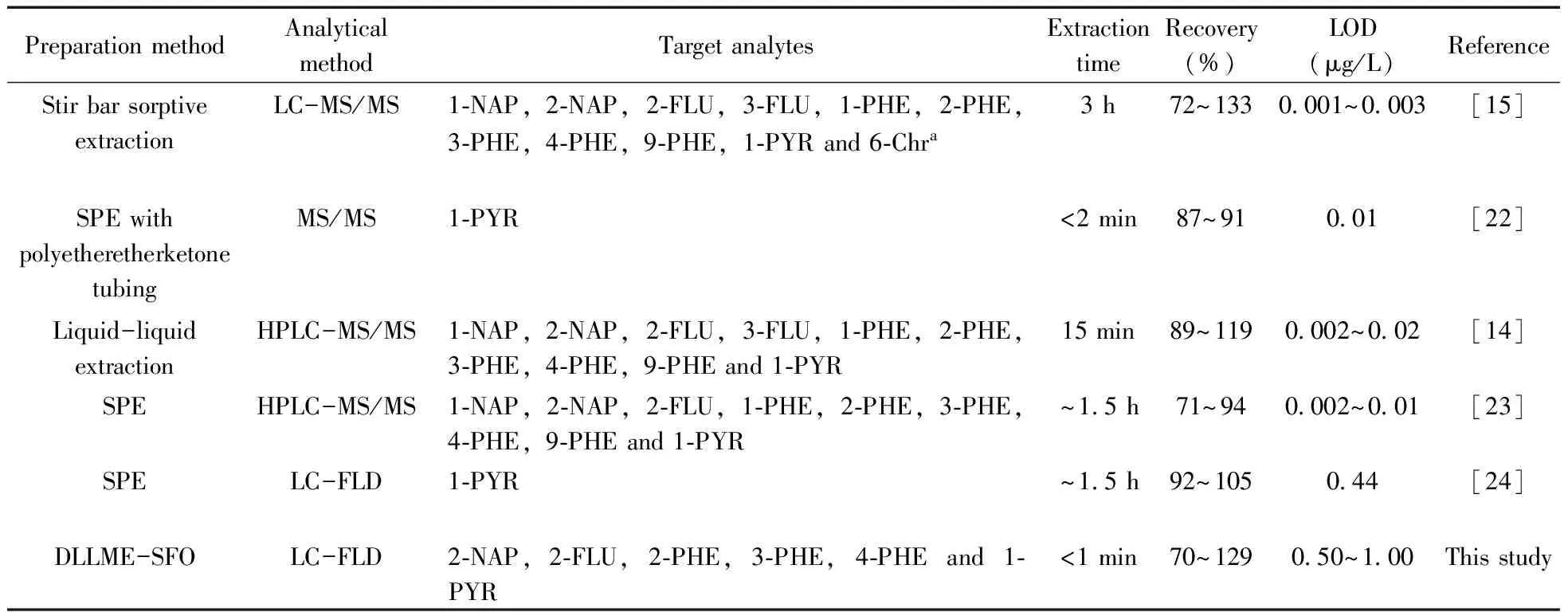
表3 本研究方法和已报道方法的比较Table 3 Comparison of various analytical methods for the detection of OH-PAHs in urine
a:6-Chr represents 6-bydroxychrysene
2.4 实际样品的检测
随机采集实验室17位志愿者的尿液样本,每个样本约10 mL,使用本方法进行检测,目标化合物的浓度使用肌酐值进行校正(表4)。除了4-PHE的检出率仅为58%,其它化合物的检出率均达到80%以上。2-NAP是最主要的检出化合物,最高浓度达到16.78 μmol/mol肌酐。证实该方法可用于人体尿液中OH-PAHs的检测。

表4 实际人体尿液中OH-PAHs的浓度Table 4 Concentrations of OH-PAHs in human urine(n=17) (μmol/mol肌酐)
3 结 论
本文建立了漂浮固化分散液液微萃取的方法,富集人体尿液中多种PAHs代谢产物,并通过液相色谱/荧光检测器(LC/FLD)进行检测。该方法显示了很好的萃取效果,操作简单快速,可满足人体尿液中6种羟基多环芳烃的分析需求,为流行病学上大样本量尿液中OH-PAHs的检测提供了参考方法。
参考文献:
[1] Kim K H,Jahan S A,Kabir E,Brown R J C.Environ.Int.,2013,60:71-80.
[2] Boffetta P,Jourenkova N,Gustavsson P.CancerCausesControl,1997,8(3):444-472.
[3] Korsh J,Shen A,Aliano K,Davenport T.BreastCare,2015,10(5):316-318.
[4] Ma Y,Harrad S.Environ.Int.,2015,84:7-16.
[5] Yang P,Wang Y X,Chen Y J,Sun L,Li J,Liu C,Huang Z,Lu W Q,Zeng Q.Environ.Sci.Technol.,2017,51(2):958-967.
[6] Yang L Y,Wang W C,Lung S C C,Sun Z L,Chen C J,Chen J K,Zou Q,Lin Y H,Lin C H.Sci.TotalEnviron.,2017,574:1649-1658.
[7] Domingo J L,Nadal M.FoodChem.Toxicol.,2015,86:144-153.
[8] Motorykin O,Santiago-Delgado L,Rohlman D,Schrlau J E,Harper B,Harris S,Harding A,Kile M L,Simonich S L M.Sci.TotalEnviron.,2015,514:170-177.
本研究采用 UPLC-MS/MS 法同时测定慢性不可预见性温和应激模型大鼠脑脊液样品中 5-羟色胺和 5-HIAA 的浓度,该方法简单、快速,可用于脑脊液中 5-羟色胺和 5-HIAA 的含量测定。
[9] Li Z,Trinidad D,Pittman E N,Riley E A,Sjodin A,Dills R L,Paulsen M,Simpson C D.J.Expos.Sci.Environ.Epidemiol.,2016,26(3):241-248.
[10] Li Z,Romanoff L C,Lewin M D,Porter E N,Trinidad D A,Needham L L,Patterson D G,Sjodin A.J.Expos.Sci.Environ.Epidemiol.,2010,20(6):526-535.
[11] Yue Q,Wang D C,Yu Z Q,Sheng G Y,Fu J M.Chin.J.Pub.Health(岳强,王德超,于志强,盛国英,傅家谟.中国公共卫生),2009,(4):443-444.
[12] Yang B C,Fang S F,Wan X J,Luo Y,Zhou J Y,Li Y,Li Y J,Wang F,Huang O P.Anal.Chim.Acta,2017,973:68-74.
[13] Wang Y,Meng L,Pittman E N,Etheredge A,Hubbard K,Trinidad D A,Kato K,Ye X,Calafat A M.Anal.Bioanal.Chem.,2017,409(4):931-937.
[14] Li Z,Romanoff L C,Trinidad D A,Hussain N,Jones R S,Porter E N,Patterson D G,Sjödin A.Anal.Chem.,2006,78(16):5744-5751.
[15] Zhao G,Chen Y,Wang S,Yu J,Wang X,Xie F,Liu H,Xie J.Talanta,2013,116:822-826.
[16] Rezaee M,Assadi Y,Milani Hosseini M R,Aghaee E,Ahmadi F,Berijani S.J.Chromatogr.A,2006,1116(1/2):1-9.
[17] Zanjani M R K,Yamini Y,Shariati S,Jönsson J Å.Anal.Chim.Acta,2007,585(2):286-293.
[18] Alshana U,Lubbad I,Goger N G,Cok I,Tamer U,Ertas N.J.Liq.Chromatogr.Relat.Technol.,2013,36(20):2855-2870.
[19] Bi H,Zheng X,Lun X W,Ma X W,Hou X H.J.Instrum.Anal.(毕欢,郑鑫,伦小文,马晓薇,侯晓虹.分析测试学报),2013,32(7):823-828.
[20] Wang Y,Zou X L,Zhang W T,Zeng H Y,Zhang L F.Chin.J.Anal.Chem.(王宇,邹晓莉,张文滔,曾红燕,张龙飞.分析化学),2013,41(5):749-753.
[21] Ahmadi-Jouibari T,Fattahi N,Shamsipur M,Pirsaheb M.J.Pharma.Biome.Anal.,2013,85:14-20.
[22] Li X,Zenobi R.Anal.Chem.,2013,85(7):3526-3531.
[23] Onyemauwa F,Rappaport S M,Sobus J R,Gajdošová D,Wu R,Waidyanatha S.J.Chromatogr.B,2009,877(11/12):1117-1125.
[24] Lu S Y,Fan R F,Yu Z Q,Sheng G Y,Fu J M.J.Liq.Chromatogr.Relat.Technol.,2012,35(11):1528-1537.
