固相萃取/超高效液相色谱-质谱法筛查及检测养鱼河水中抗生素
2018-04-18程家兴赵起越李令军杜振霞郭巧珍沈正超
程家兴,赵起越,李令军,杜振霞*,郭巧珍,3,沈正超
(1.北京化工大学 理学院,北京 100029;2.北京市环境保护检测中心,北京 100048;3.北京市疾病预防控制中心 北京市食物中毒溯源诊断重点实验室,北京 100013)
养殖业普遍存在以兽用抗生素治疗各种因细菌真菌感染产生的疾病的现象[1],部分企业甚至在动物饲料中直接添加兽用抗生素以达到增产的目的[2-3]。2012年美国和韩国开始规范饲料中抗生素的使用[4],但我国并没有严格的立法限制,导致养殖业滥用抗生素问题严重[5]。水产养殖直接使用抗生素会导致周围水环境受到污染,并且抗生素在不同环境中的化学行为也不一致[6]。Pomati等[7]的研究结果显示,水体中四环素或红霉素等抗生素会严重抑制淡水单细胞藻类的生长。常用的氯霉素可引起再生障碍性贫血,大环内酯类、磺胺类等具有抗原性,可引起人体的过敏反应[8],直接影响人类以及其它动植物用水安全。
抗生素的常用检测方法有高效液相色谱-紫外/荧光(HPLC-DAD/FLD)[9-10]法、液相色谱-串联质谱(LC-MS/MS)[11-13]法等。有文献报道[14]使用超高效液相色谱-四极杆飞行时间质谱法(UPLC-Q-TOF MS)对牛奶中的抗生素进行分析,但无法检测到实际样品中的痕量物质(ng/L)。
而对北方地区养鱼业中抗生素的使用情况及其对周围地表水环境影响的研究,目前几乎没有相关文献。本实验首次使用UPLC-Q-TOF MS结合UPLC-MS/MS对养鱼河水中抗生素进行快筛和痕量分析(ng/L),弥补了TOF灵敏度不高、定量准确度较差,以及三重四极杆质谱MRM模式只能对目标化合物进行分析的缺陷。与地下水及自来水相比,养鱼河水成分复杂,基质效应更加严重。与其他文献报道[15-17]相比,本实验着重优化了水样的前处理条件,在富集净化4大类15种抗生素的同时具备较高的回收率及较小的基质效应。
1 实验部分
1.1 仪器与试剂
Waters Acquity超高效液相色谱仪、Waters Xevo TQ-XS 三重四极杆质谱仪、Waters Xevo G2-S 四极杆飞行时间质谱(美国Waters 公司);12位防交叉污染固相萃取仪(美国Supelco公司);MTN-2800D 12位手动氮吹仪(天津奥特赛恩斯仪器有限公司);FE20 pH 计(上海梅特勒-托利多仪器有限公司);Oasis HLB固相萃取柱(500 mg/6 mL,美国 Waters公司)。

甲醇、乙腈为色谱纯(美国 Fisher 公司);甲酸为色谱纯(赛默飞世尔科技(中国)有限公司);盐酸为分析纯(北京化工厂);乙二胺四乙酸二钠(Na2EDTA)为分析纯(国药集团化学试剂有限公司)。
0.45 μm玻璃纤维滤膜、0.22 μm尼龙滤膜(天津津腾实验有限公司)。
1.2 标准溶液配制
准确称取1 mg(精确至0.01 mg) 15种抗生素标准品,用含0.1%甲酸的甲醇配制成质量浓度为1 g/L的标准溶液保存于-20 ℃的冰箱中,使用时以初始流动相稀释。
1.3 样品采集
在养鱼池及其附近河流设置采集点,每个采样点采集2.5 L河水于褐色玻璃瓶中密封避光保存,在4 ℃下冷藏,24 h内完成测定。
1.4 样品前处理
1.4.1水样品过滤在室温下,水样用0.45 μm玻璃纤维滤膜过滤后准确量取500 mL于1 L玻璃烧杯中,用盐酸调至pH 4.0,加入0.5 g Na2EDTA,充分混匀,待上柱净化、富集。
1.4.2固相萃取依次用6.0 mL甲醇、6.0 mL超纯水(pH 4.0)活化HLB固相萃取柱。样品以约3 mL/min的流速通过柱子,用6 mL超纯水淋洗,真空泵抽干。干燥后,分别用4 mL甲醇和4 mL含0.1%甲酸的甲醇洗脱。将洗脱液收集于10 mL离心管中,氮气吹扫至近干,然后用初始流动相复溶至0.50 mL,过0.22 μm滤膜,待上机测定。
1.5 仪器条件
1.5.1色谱条件色谱柱:BEH C18(100 mm×2.1 mm,1.7 μm,美国 Waters 公司);进样量:2 μL;柱温:40 ℃;流动相:A为甲醇;B为 0.1%(体积分数)甲酸水溶液。
ESI正离子模式梯度洗脱程序(流速0.35 mL/min):0~1.00 min,5%A;1.00~2.00 min,5%~20%A;2.00~6.50 min,20%~34%A;6.50~7.00 min,34%~95%A;7.00~7.50 min,95%A;7.50~8.00 min,95%~100%A;8.00~8.30 min,100%A;8.30~8.40 min,100%~5%A;8.40~10.00 min,5%A。
ESI负离子模式梯度洗脱程序(流速0.30 mL/min):0~1.00 min,5%A;1.00~2.00 min,5%~40%A;2.00~5.00 min,40%~45%A;5.00~5.10 min,45%~95%A;5.10~5.50 min,95%~5%A;5.50~7.00 min,5%A。
1.5.2飞行时间质谱仪条件电离模式:ESI+/ESI-;检测模式:MSE;毛细管电压:3 kV;离子源温度:120 ℃;脱溶剂气温度:450 ℃;脱溶剂气流速:800 L/h;锥孔电压:30 V,碰撞能量20~40 eV;LockSpray:m/z556.2776,每30 s切换一次进行质量数校正。
1.5.3三重四极杆质谱仪条件电离模式:ESI+/ESI-;检测方式:多反应监测(MRM)模式。优化后的质谱条件见表1。
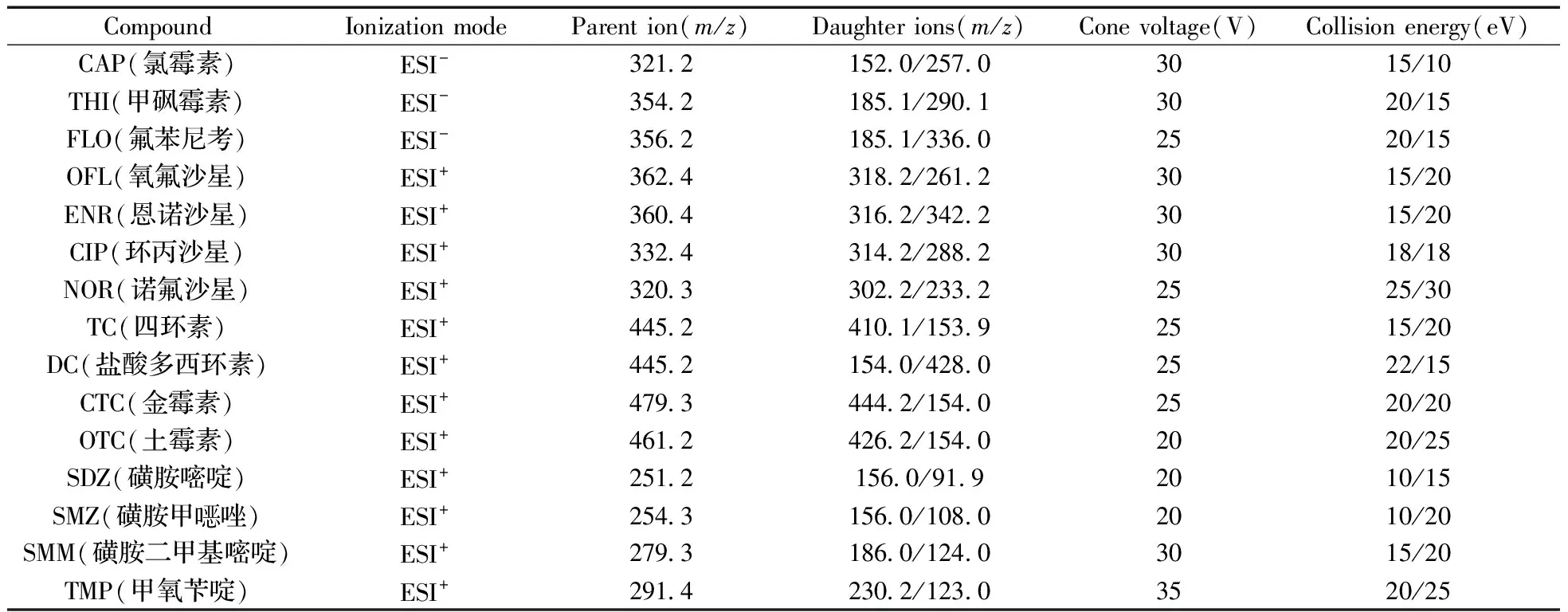
表1 15种抗生素的质谱条件Table 1 MS/MS conditions of the 15 antibiotics
2 结果与讨论
2.1 前处理条件优化
2.1.1固相萃取柱的选择结合文献[13,18]的报道,本实验比较了HLB固相萃取柱和Sep-Pak C18固相萃取柱的效果。实验结果显示,后者对四环素类、磺胺类的回收率均远低于前者,例如与Sep-Pak C18固相萃取柱相比,TC在HLB固相萃取柱上回收率提高了30%,SDZ在HLB固相萃取柱上回收率提高了21%。而对于氯霉素类和喹诺酮类抗生素,HLB固相萃取柱和Sep-Pak C18固相萃取柱的回收率相差不大。
进一步对6 mL/500 mg和3 mL/60 mg两种规格的HLB固相萃取柱进行对比,发现6 mL/500 mg对磺胺类的回收率更高,而且与3 mL/60 mg 相比TC回收率从原来的45%提高到65.3%,而DC则从原来的70%提高到82.5%。两种规格HLB柱对喹诺酮类和氯霉素类的回收率无太大差异。原因是上样体积大时3 mL/60 mg规格的固相萃取柱容易过载。因此选用体积容量较大的6 mL/500 mg的HLB固相萃取小柱。
2.1.2水样pH值的调节部分抗生素样品在碱性环境中不能稳定存在,用盐酸调节样品pH值至2.0和4.0,分别比较原样品与pH 2.0和pH 4.0时的回收率。将pH值调至4.0时,磺胺类的回收率可从不调节pH值的61.2%增至70.9%,四环素类和喹诺酮类的回收率也均有明显提高,而当pH值调至2.0时,一些喹诺酮类抗生素的回收率略有下降。pH值对氯霉素类抗生素的影响不大。综合考虑,本实验选择调节水样品pH值为4.0。
2.1.3上样体积的选择实验比较了500 mL和1 000 mL上样体积的回收率,发现使用500 mL上样体积时,氯霉素类和磺胺类抗生素的回收率略有提高,喹诺酮类和四环素类抗生素的回收率明显提高,例如上样体积从1 000 mL减少至500 mL时,NOR从30%提高到73.6%。上样体积太大可能使水样品中杂质吸附在固相萃取柱填料上或超载,导致喹诺酮类和四环素类抗生素回收率下降,因此实验选用的上样体积为500 mL。
2.1.4洗脱溶液的选择实验比较了纯甲醇和含0.1%甲酸甲醇的洗脱效果。结果显示,以纯甲醇洗脱时,DC、TC、NOR、ENR的回收率比用0.1%甲酸甲醇洗脱时平均低10%左右。但纯甲醇洗脱时CIP和OTC的回收率略高于0.1%甲酸甲醇洗脱时的回收率。氯霉素类和磺胺类抗生素的回收率则差别不大。综合考虑使用纯甲醇和0.1%甲酸甲醇分别进行洗脱,以提高回收率。
2.1.5洗脱体积的选择分别选用3 mL甲醇和3 mL 0.1%甲酸甲醇、4 mL甲醇和4 mL 0.1%甲酸甲醇、5 mL甲醇和5 mL 0.1%甲酸甲醇进行洗脱,发现4 mL甲醇和4 mL 0.1%甲酸甲醇与5 mL甲醇和5 mL 0.1%甲酸甲醇的回收率差别不大,且对喹诺酮类和磺胺类抗生素的回收率均高于3 mL甲醇和3 mL 0.1%甲酸甲醇,例如使用3 mL甲醇和3 mL 0.1%甲酸甲醇洗脱时OFL的回收率为59%,使用4 mL甲醇和4 mL 0.1%甲酸甲醇洗脱时回收率提高到70.6%。故选用4 mL甲醇和4 mL 0.1%甲酸甲醇进行洗脱处理。
2.2 基于UNIFI软件建立抗生素数据库
利用ChemSpider专业化学数据库整理生活中常用的103种抗生素的mol文件,将其导入UNIFI软件中。建立包含名称、分子式、结构式、精确质量数、特征离子、中性丢失、可能的MS/MS裂解规律等信息的 UNIFI抗生素数据库。利用该数据库对经UPLC-Q-TOF MS分析后的水样数据进行快速筛查。筛查条件为质量偏差小于10 mDa。由于缺少全部的抗生素标准品在色谱柱的保留时间数据,在筛查完成后,对筛查到的抗生素,购买标准品后通过三重四极杆质谱进行准确的定性定量分析。
2.3 仪器条件的优化
2.3.1色谱条件优化甲酸是在ESI+模式下增强被测物质离子化程度的常用试剂,同时能够有效改善峰形,使峰形更加尖锐。因此选择含0.1%甲酸的水溶液作为流动相B。由于本实验的抗生素用甲醇溶液配制,因此选用甲醇作为流动相A。
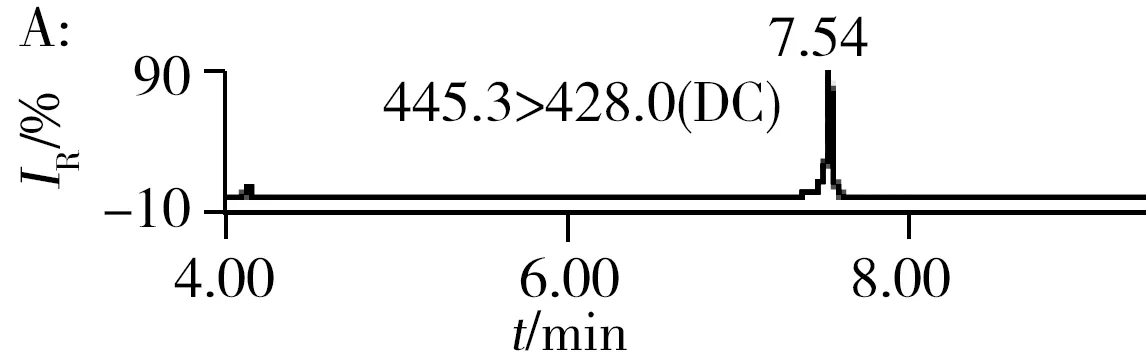
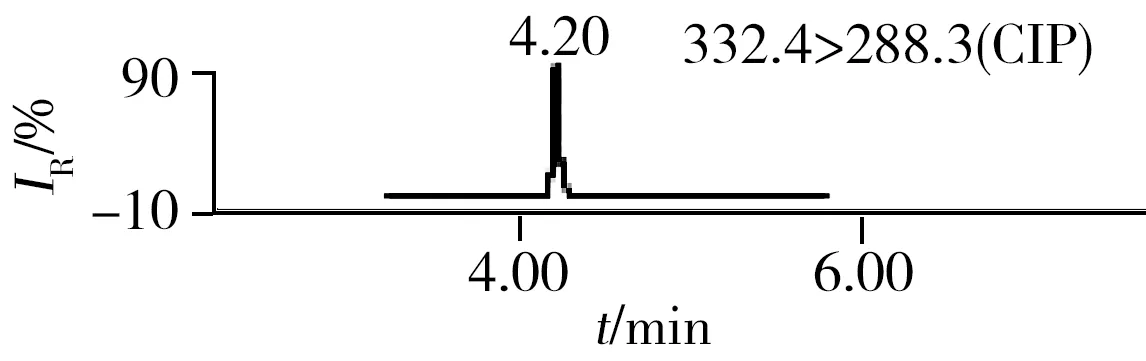
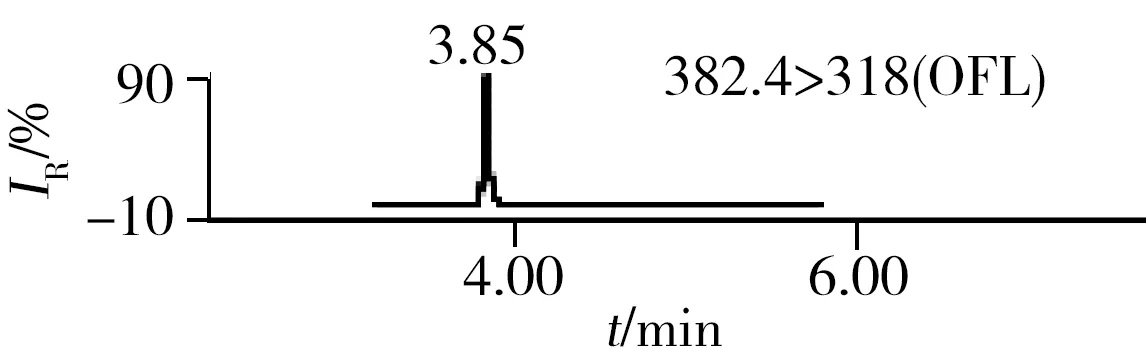
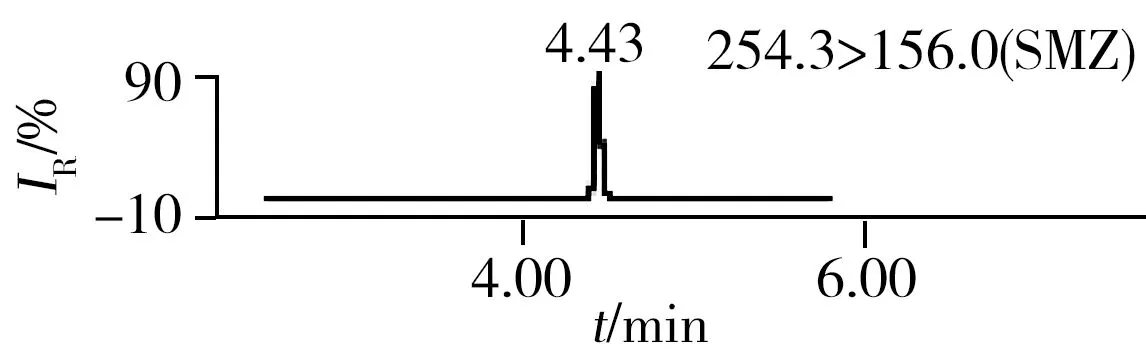
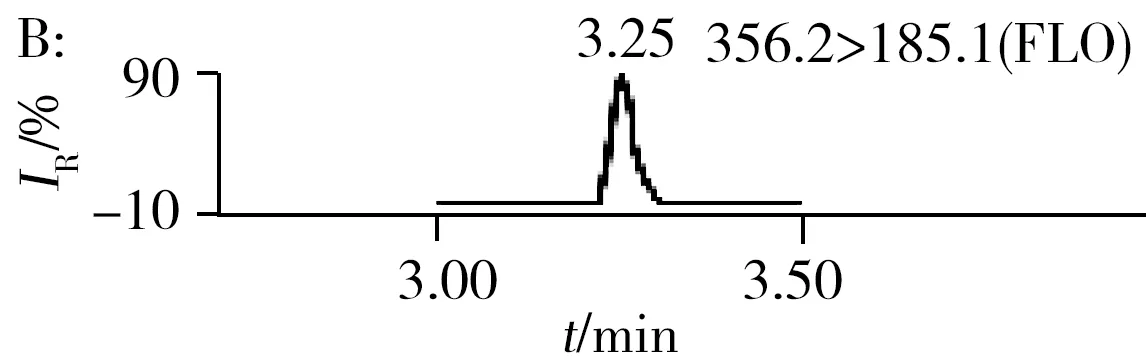
2.3.2三重四极杆质谱条件优化抗生素标准品在全扫描模式下直接进样,以从小到大的方式改变锥孔电压,由产生的峰高判断每种抗生素最合适的锥孔电压并确定母离子的相对分子质量,经子离子扫描选择最优的两对子离子的质量数及最佳碰撞能量。为了进一步提高反应的灵敏度,采用多时段多离子反应监测模式。15种抗生素的色谱图如图1所示。
2.4 线性范围与检出限
将“1.2”配制的标准溶液用初始流动相稀释成一系列不同质量浓度的溶液,以质量浓度为横坐标(x,μg/L),峰面积为纵坐标(y),绘制15种抗生素的标准曲线(表2)。除了金霉素的线性范围为1.0~50 μg/L,其余抗生素均在0.03~50 μg/L范围内线性良好,相关系数r≥0.990。在空白水样中添加15种抗生素,经过“1.4”前处理方法后得到不同抗生素的方法检出限(MDL,S/N≥3),见表2。15种抗生素的检出限为0.01~0.3 ng/L。
将本方法与其他文献报道的方法进行比较(表3),结果表明,相较于HPLC,UPLC在保证抗生素分离效果的情况下所需分析时间更短。相同柱长下,本实验的分析时间较短。本方法对各类抗生素的检出限均低于或接近其他方法的检出限,且灵敏度较高。
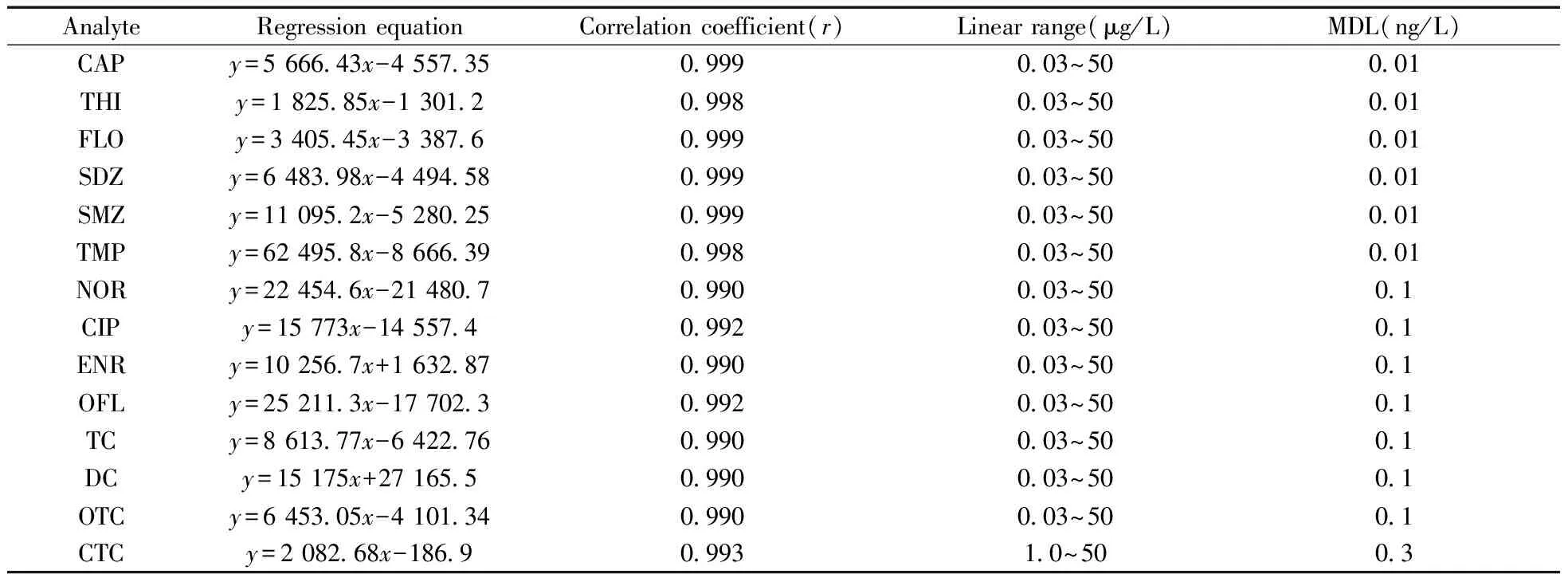
AnalyteRegressionequationCorrelationcoefficient(r)Linearrange(μg/L)MDL(ng/L)CAPy=5666 43x-4557 350 9990 03~500 01THIy=1825 85x-1301 20 9980 03~500 01FLOy=3405 45x-3387 60 9990 03~500 01SDZy=6483 98x-4494 580 9990 03~500 01SMZy=11095 2x-5280 250 9990 03~500 01TMPy=62495 8x-8666 390 9980 03~500 01NORy=22454 6x-21480 70 9900 03~500 1CIPy=15773x-14557 40 9920 03~500 1ENRy=10256 7x+1632 870 9900 03~500 1OFLy=25211 3x-17702 30 9920 03~500 1TCy=8613 77x-6422 760 9900 03~500 1DCy=15175x+27165 50 9900 03~500 1OTCy=6453 05x-4101 340 9900 03~500 1CTCy=2082 68x-186 90 9931 0~500 3
y:peak area of the target compound;x:mass concentration(μg/L)
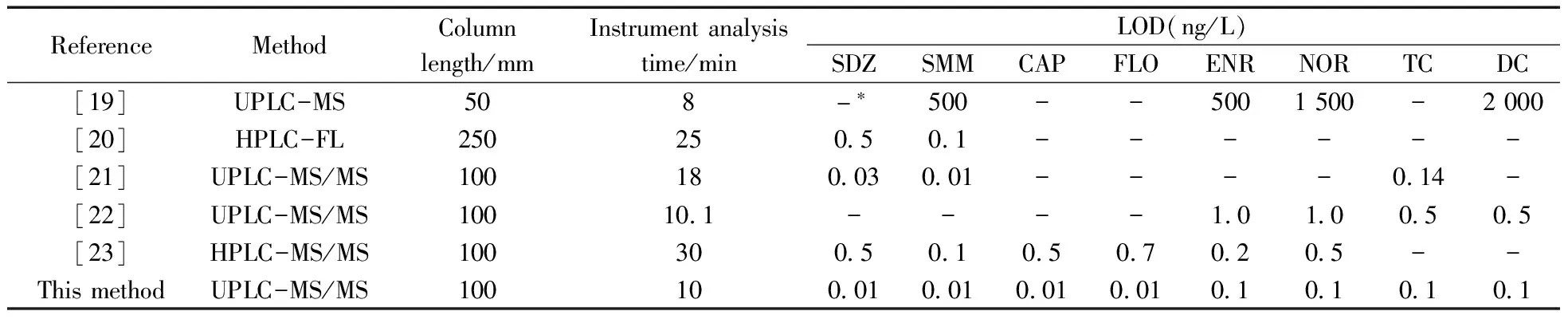
表3 本方法与其他方法的比较Table 3 Comparison of this method with other methods
*untested
2.5 回收率与精密度
用河水上游的500 mL水库水作为空白水样,分别添加10、30 ng 的1 g/L混合标准溶液,每个加标水平做3个平行,结果见表4。结果表明,15种抗生素的回收率为61.0%~98.4%,相对标准偏差(n=3)为4.6%~14.0%,日内和日间的相对标准偏差分别为6.8% ~ 8.7%和7.2% ~9.8%。
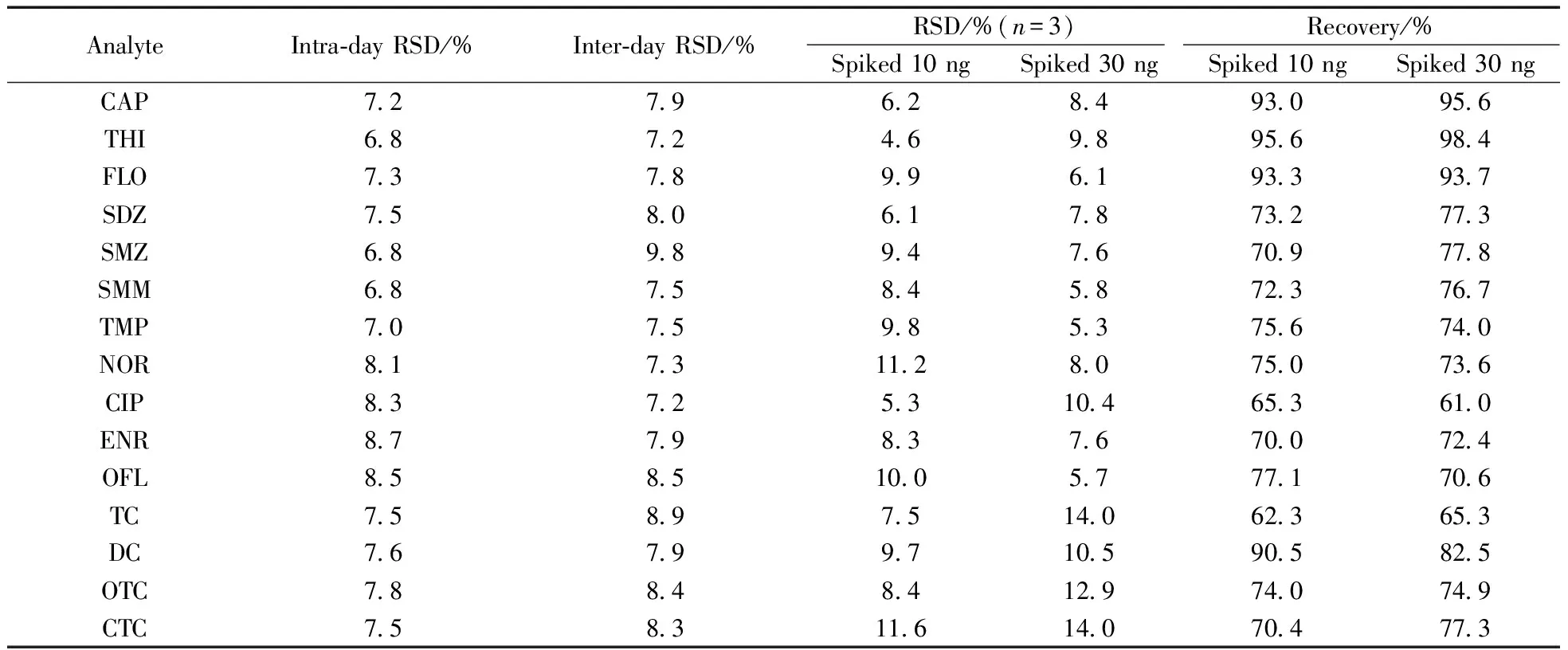
表4 15种抗生素的平均加标回收率和相对标准偏差(n=3)Table 4 Average spiked recoveries and relative standard deviation(RSDs) for the 15 antibiotics at 2 spiked levels(n=3)
2.6 实际样品分析
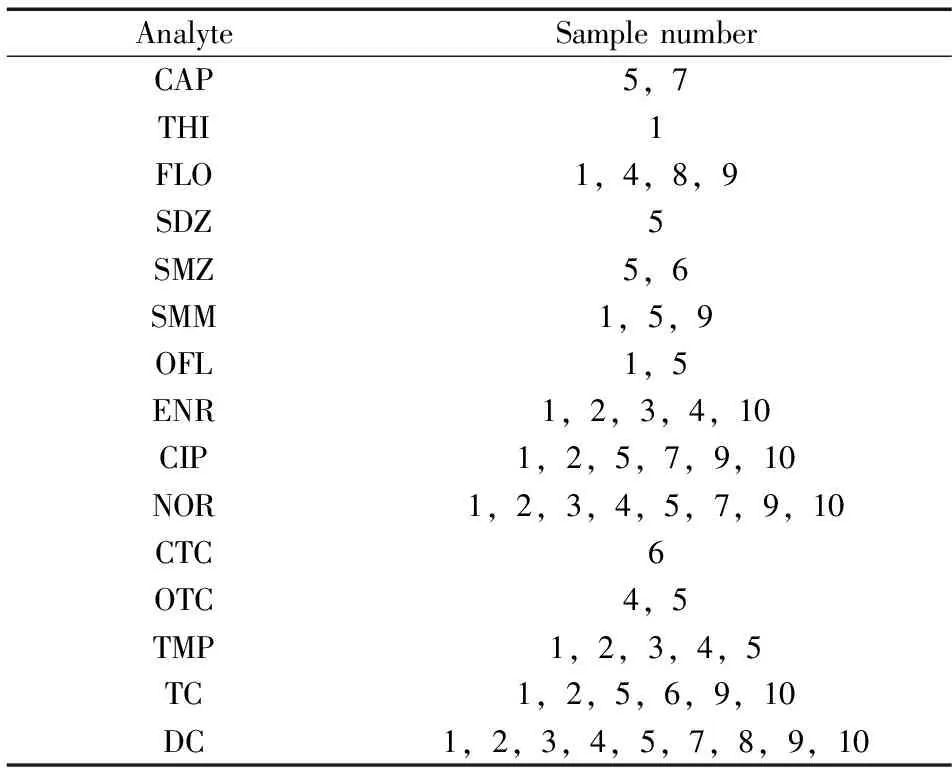
表5 水样的初筛结果Table 5 The detection data of UPLC-TOF MS
the sample numbers were the same as those in Table 6
2.6.1利用超高效液相色谱串联四极杆飞行时间质谱的快筛利用本研究建立的UPLC-Q-TOF MS方法对10个采样点的水样进行了分析,水样经过“1.4”方法前处理后,用建立的抗生素数据库对分析结果进行快筛,结果如表5,样品编号与表6采样点编号相同。
2.6.2实际样品的定性定量分析利用本研究建立的UPLC-MS/MS方法对10个采样点的水样进行了检测,结果表明,氯霉素类和喹诺酮类相较于其它类抗生素的检出率更高,而且在部分采样点检出的浓度较高(表6)。对比表5,发现初筛的结果准确性较高,初筛的15种抗生素在实际样品中均有检出,其中含量较低的抗生素使用灵敏度较高的三重四极杆质谱才能准确定性定量。
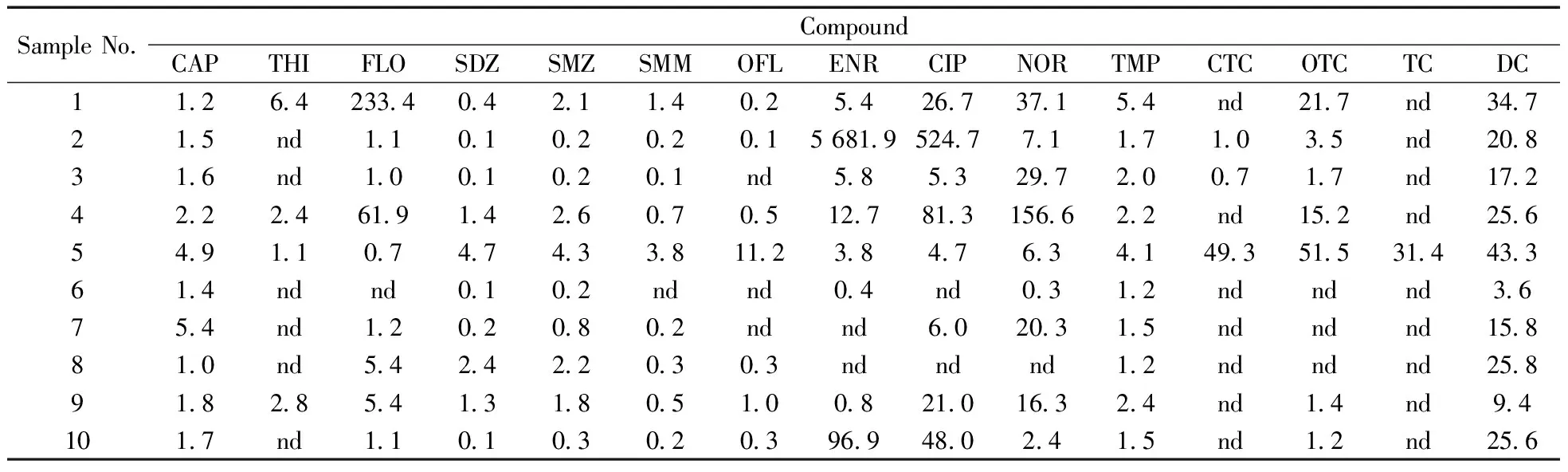
表6 10个采样点的检测数据Table 6 The detection data of 10 sampling points (ng/L)
nd:lower than the LOD
3 结 论
有关北方地区水产养殖中兽用抗生素的使用情况研究较为缺乏,本研究前期结合UNIFI软件建立的数据库使用UPLC-Q-TOF MS对水样中的抗生素进行快速筛查,有效避免了使用三重四极杆质谱检测抗生素的盲目性。再使用UPLC-MS/MS的MRM模式进一步准确定性和定量养鱼河水中筛查到的4类(喹诺酮类、氯霉素类、四环素类、磺胺类)15种抗生素,其中氯霉素类和喹诺酮类抗生素的检出率与检出浓度较高。优化之后的固相萃取方法有效避免了样品的基质效应。该方法灵敏度较高,重复性好,可在10 min内快速检测河流等复杂水环境中的抗生素。
参考文献:
[1] Yan L Y,Liu G H,Qin S,Hu G,Fan C W.ActaAgric.Jiangxi(严莲英,刘桂华,秦松,胡岗,范成五.江西农业学报),2016,28(9):90-94.
[2] CEC.1998a.Council Regulation 2788/98.OfficialJournaloftheEuropeanCommunitiesLegislation,L347,323.
[3] CEC.1998b.Council Regulation 2821/98.OfficialJournaloftheEuropeanCommunitiesLegislation,L351,4.
[4] Guo X Y,Wang N,Xu J,Hao L J,Zhi Y,Wang Z C,Shan Z J.Environ.Chem.(郭欣妍,王娜,许静,郝利君,智勇,王昝畅,单正军.环境科学与技术),2014,(9):76-86.
[5] Lei T,Ping L,Wang Y X,Zhu K Z.Chemosphere,2009,74(8):1090-1097.
[6] Managaki S,Murata A,Takada H,Tuyen B C,Chiem N H.Environ.Sci.Technol.,2007,41(23):8004-8010.
[7] Pomati F,Netting A G,Calamari D,Neilan B A.Aquat.Toxicol.,2004,67(4):387-396.
[8] Qi Y J,Liu F.RockMiner.Anal.(祁彦洁,刘菲.岩矿测试),2014,33(1):1-11.
[9] Renew J E,Huang C H.J.Chromatogr.A,2004,1042(1):113-121.
[10] Sun P,Barmaz D,Cabrera M L,Pavlostathis S G,Huang C H.J.Chromatogr.A,2013,1312(17):10-17.
[11] Yan C,Yang Y,Zhou J,Liu M,Nie M H,Shi H,Gu L J.Environ.Pollut.,2013,175(8):22-29.
[12] Bao X L,Ren Y P,Zhang H.Chin.J.Anal.Chem.(包晓丽,任一平,张虹.分析化学),2009,37(3):389-394.
[13] Wu D,Han M L,Zou D X,Wang X M,Gao M,Chou T L.Chin.J.Anal.Chem.(吴丹,韩梅琳,邹德勋,王旭明,高敏,仇天雷.分析化学),2017,(9):1389-1396.
[14] Zhang J,Yan L J,Pan C S,Lin L Y,Zhang X Y,Shen H Q.Chin.J.Chromatogr.(张洁,严丽娟,潘晨松,林立毅,张欣怡,申河清.色谱),2012,30(10):1031-1036.
[15] Li X J,Pan Y X,Yu H,Gan P S,Zhu H Y.Chin.J.HealthLab.Technol.(李晓晶,潘幼娴,于鸿,甘平胜,朱惠扬.中国卫生检验杂志),2013,(11):2426-2428.
[16] Zhang X Y,Li Z Q,Li L C,Liu Y L.J.ZhongzhouUniv.(张晓燕,李志强,李领川,刘艳丽.中州大学学报),2013,30(4):114-118.
[17] Wang J L,Xu J J,Cai Z X,Zhang N H,Wang Z Y,Ren Y P.Chin.J.HealthLab.Technol.(王军淋,许娇娇,蔡增轩,张念华,王志园,任一平.中国卫生检验杂志),2016,(17):2443-2449.
[18] Cai Z B,Zhang Y,Liu L.Chin.J.HealthLab.Technol.(蔡志斌,张英,刘丽.中国卫生检验杂志),2007,17(2):270-272.
[19] Li L,Zhang Y L,Lin T,Wang J L,Liu H C.J.Instrum.Anal.(李宁,张玉龙,林涛,王继良,刘宏程.分析测试学报),2016,35(6):714-718.
[20] Wu C Q,Lei J M,Li Y L,Wang Y L,Chen D Y,Gong J.Chin.J.Chromatogr.(吴翠琴,雷金妹,李韵灵,王韵靓,陈迪云,龚剑.色谱),2014,32(12):1362-1367.
[21] Wang S,Zhang X M,Zhang J,Shao B,Li S M.Chin.J.Chromatogr.(王硕,张向明,张晶,邵兵,李书明.色谱),2015,33(7):730-739.
[22] Wang M J,Hu J W,Tian L,Qiao H O,Nie X L.Chin.J.HealthLab.Technol.(王敏娟,胡佳薇,田丽,乔海鸥,聂晓玲.中国卫生检验杂志),2017,(4):473-476.
[23] Du J,Zhao H X,Chen J W.Chin.J.Chromatogr.(杜鹃,赵洪霞,陈景文.色谱),2015,33(4):348-353.
