双羰基氰吡咯化合物的电子吸收光谱及其溶剂效应的理论研究❋
2018-04-13张广龙修方圆赵思琪夏树伟于良民
张广龙, 修方圆, 赵思琪, 夏树伟, 于良民
(中国海洋大学海洋化学理论与工程技术教育部重点实验室,化学化工学院,山东 青岛 266100)
吡咯是重要的含氮五元杂环分子,在进行特定基团的修饰后具有良好的荧光性质[1-3],其中双羰基吡咯衍生物因其较强的共轭结构而备受关注[4]。近年来,利用密度泛函及含时密度泛函对分子结构及电子光谱[5-11]的计算越来越普遍,可获得分子从基态到激发态跃迁的路径、能量等信息,尤其能直接获得溶剂效应对分子微观结构及光谱行为的影响,为筛选荧光分子提供了参考和理论支持,有助于新产品的设计和研发。
本文利用密度泛函方法,对双羰基氰吡咯化合物及其在苯、甲苯、氯仿、四氢呋喃及乙醇溶剂中的结构和电子光谱进行了计算,分析了溶剂的极性对分子结构和电子光谱的影响,为深入研究溶剂效应对分子结构和光谱行为的影响提供理论基础。
1 计算方法
双羰基氰吡咯化合物分子结构如图1所示,在DFT的B3LYP/6-31G(d,p)水平上,对目标分子分别在真空和溶剂中的几何构型进行优化,并对优化构型进行频率分析确认。在此基础上用TD-DFT的B3LYP/6-31G(d,p)计算得到电子吸收光谱。使用自洽反应场(SCRF)方法,并结合极化连续介质模型(PCM)衡量溶剂场效应。全部计算使用Gaussian 09程序包[12]。
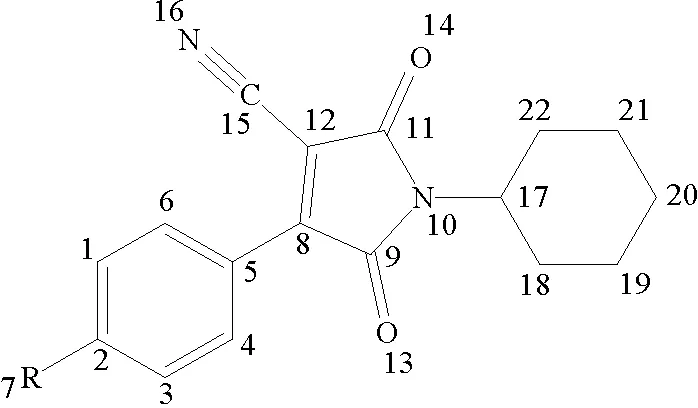
图1 分子结构式
2 结果与讨论
2.1 溶剂极性对双羰基氰吡咯化合物几何构型的影响
分别对双羰基氰吡咯化合物分子在真空、苯、甲苯、氯仿、四氢呋喃及乙醇溶剂中的几何构型进行优化,其结构参数(键长、键角)和能量等列于表1。
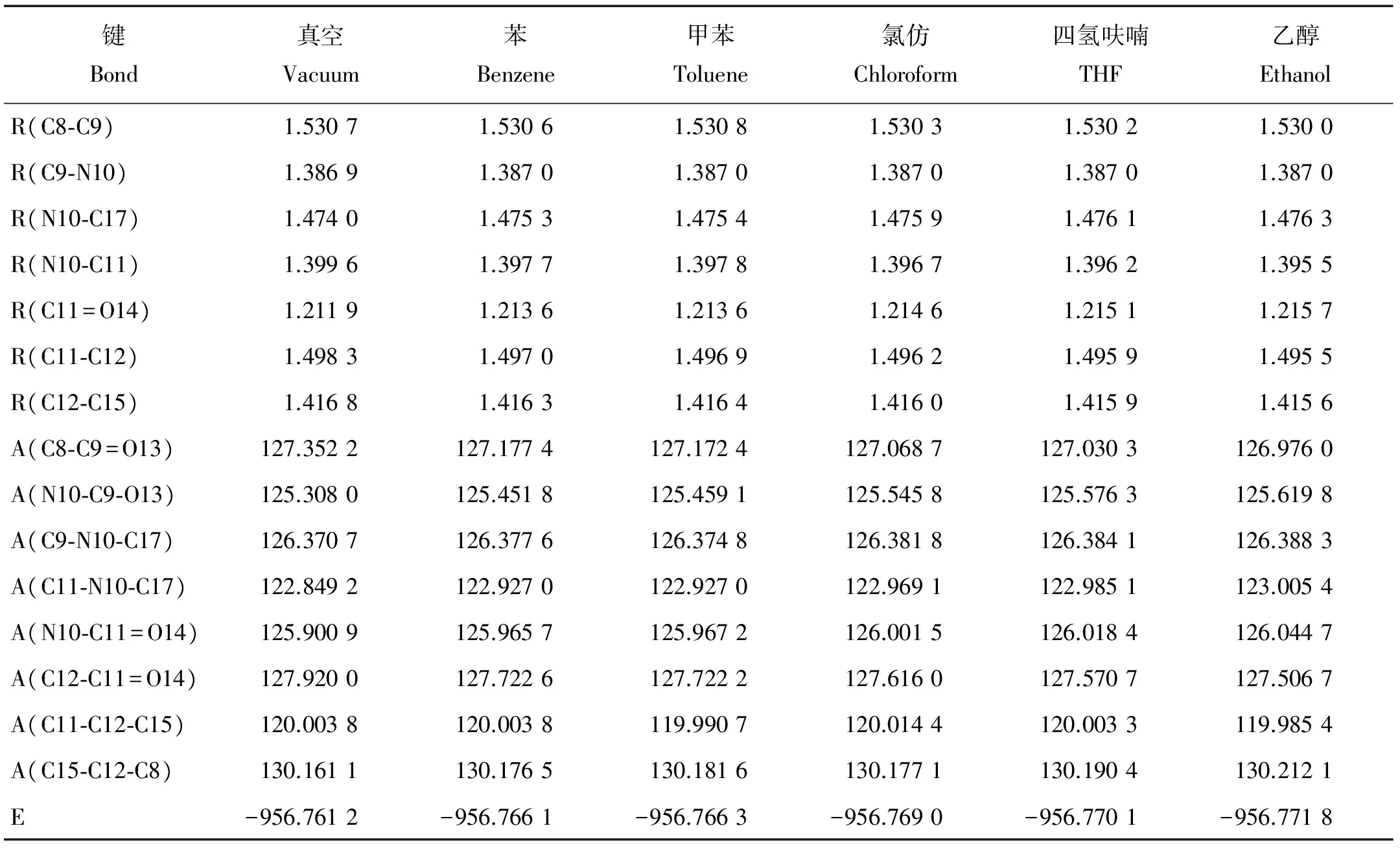
表1 双羰基氰吡咯分子在真空及不同溶剂中平衡构型的结构参数
注:R: 键长(Å),A: 键角(°),E: 总能量(Hartree)。R:Bond distance(Å),A:Bond angle(°),E:Total energy(Hartree).
由表1中的键长和键角看出,不同溶剂对分子构型的影响主要体现在吡咯环上的N10和C11,除C9-N10键长外,其余键长均随溶剂的极性增强而有不同程度的变化。与真空下的分子结构相比,在苯、甲苯、氯仿、四氢呋喃及乙醇溶剂(介电常数:苯<甲苯<氯仿<四氢呋喃<乙醇)中,N10-C17单键分别增加了0.001 3,0.001 4,0.001 9, 0.002 1,0.002 3Å;C11=O14双键分别增加了0.001 7,0.001 7,0.002 7,0.003 2,0.003 8Å;N10-C11单键分别减小了0.001 9,0.001 8,0.002 9,0.003 4,0.004 1Å;C11-C12单键分别减小了0.001 3,0.001 4,0.002 1,0.002 4,0.002 8Å。可见,键长增加或减小的程度有随着溶剂极性的增加而增大的趋势。
同样,分子的键角也随着溶剂极性的不同而改变。键角∠N10-C9-O13分别增大了0.143 8°,0.151 1°,0.237 8°,0.268 3°,0.311 8°;键角∠C8-C9=O13分别减小了0.174 8°, 0.179 8°,0.283 5°,0.321 9°,0.376 2°;键角∠C12-C11=O14键角分别减小了0.197 4°,0.197 8°,0.304 0°,0.349 3°,0.413 3°。由此可见,随着溶剂极性的增加双羰基氰吡咯化合物分子结构发生较有规律的变化,结构的改变使电子光谱发生一定程度的变化[13]。溶剂的加入对体系总能量的影响较为明显,随着溶剂极性的增大,分子体系总能量有逐渐降低的趋势。
2.2 前线分子轨道
前线分子轨道HOMO和LUMO及其能级差△EH-L与光谱特性密切相关[14-15]。其△EH-L越大,说明分子越难被激发产生电子光谱。在不同溶剂中分子的前线轨道分布相似(以在真空和乙醇中为例,见图2)。可以看出,前线轨道主要分布在芳环和吡咯环上,电子从HOMO轨道跃迁到LUMO属于π→π*跃迁。真空下分子的C3,C5,C6,N10,C12对最高占据轨道的贡献率分别为15.60%,5.20%,16.50%,8.02%,19.50%;而在乙醇溶剂中的分别为16.39%,4.85%,17.74%,7.05%,19.00%;真空下分子的C3,C8,C9,C12,O13,O14对最低空轨道的贡献率分别为5.72%,23.20%,7.50%,16.70%,10.78%,10.29%。在乙醇溶剂中的分别为5.45%,24.07%,7.82%,15.10%,11.12%,10.02%。通过对比分析最高占据轨道和最低空轨道的组成可知,在真空和乙醇溶剂中略有差异,其他溶剂也是如此,因此溶剂对分子的电子结构有一定的扰动,导致其电子光谱的差异。
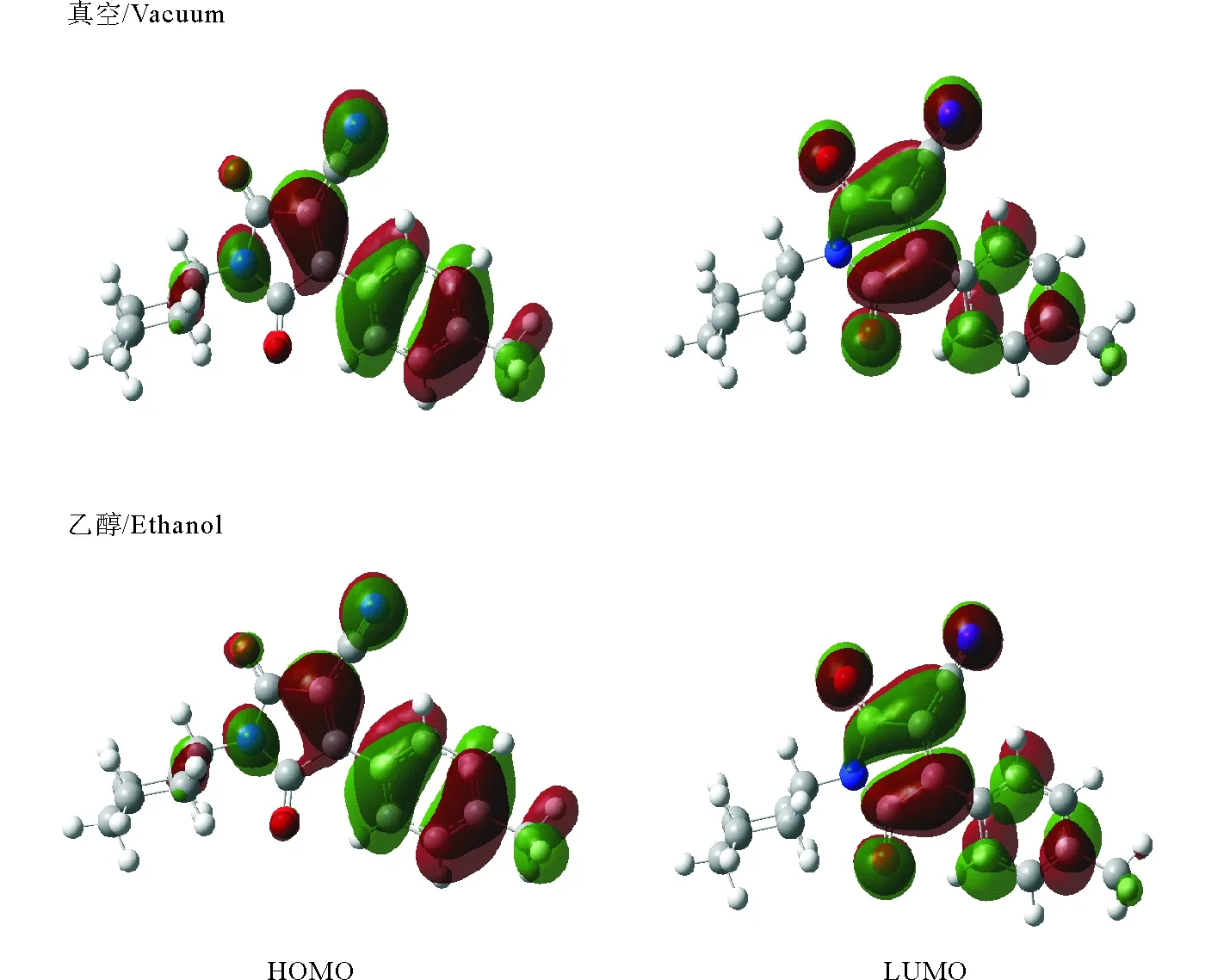
图2 双羰基氰吡咯化合物分子在真空和乙醇溶剂中HOMO和LUMO轨道图
2.3 溶剂的极性对电子光谱影响
电子光谱能够反映化合物基态与激发态能级间的跃迁情况。采用含时密度泛函计算了分子在真空及不同溶剂中的电子吸收光谱(见图3),得到了跃迁能、主要跃迁贡献、最大吸收波长和振子强度等光谱特征参数(见表2)。
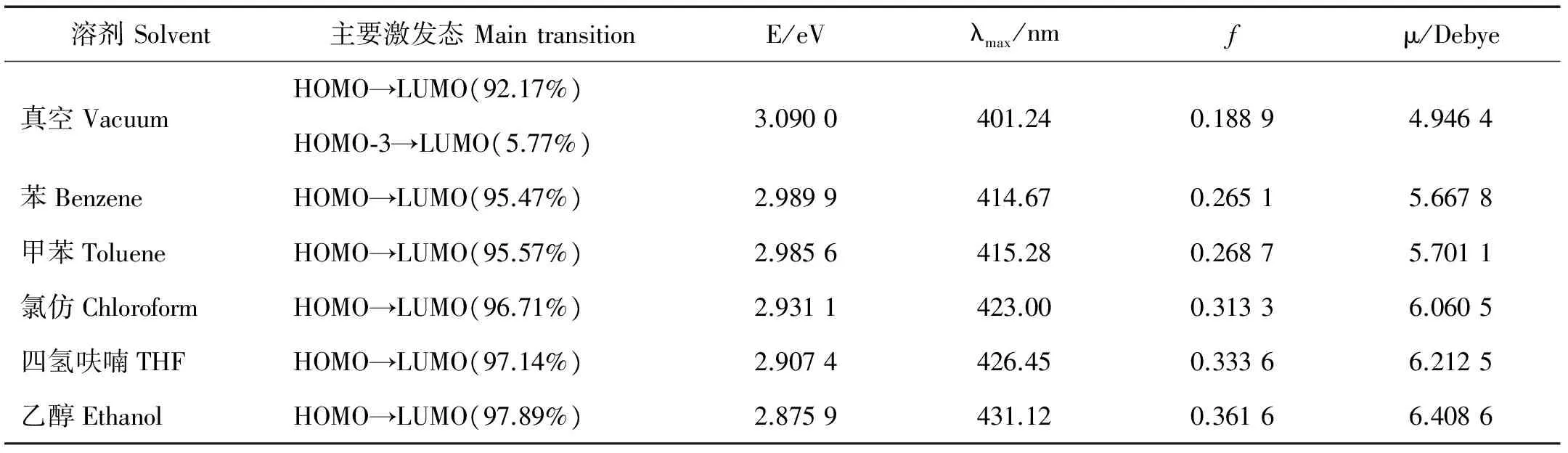
表2 分子在真空及不同溶剂中的电子吸收光谱参数
显然,第一激发态主要来自电子从HOMO到LUMO的跃迁,除此以外HOMO-3或HOMO-2到LUMO的跃迁也有参与,但贡献不足6%。溶剂对分子跃迁能级差的影响体现在最大吸收波长的移动,从表2看出,最大吸收波长在加入溶剂后,均产生不同程度的红移,λmax随溶剂极性增强而增大,由401 nm增大到431 nm,且吸收峰的强度也逐渐增大。这是因为,极性溶剂对分子激发态具有稳定作用,且极性增强其稳定作用程度增大,而对分子基态影响小,因此电子跃迁能量下降,发生吸收波长的红移。另外,分子的偶极矩也随着溶剂极性增强而增大,因此吸收峰的强度的变化趋势与之一致。
3 结论
利用密度泛函理论B3LYP/6-31G (d, p)方法结合PCM溶剂化模型,计算了真空和五种溶剂中双羰基氰吡咯化合物分子的平衡结构和电子吸收光谱,分析了溶剂对分子结构和电子吸收光谱的影响。主要得出以下几点结论:
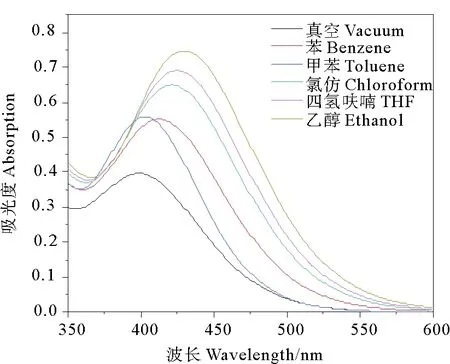
图3 在真空及不同溶剂中的紫外-可见吸收光谱图
(1)极性溶剂对分子结构的影响主要体现在吡咯环和杂原子上,且溶剂的极性越强,分子结构的变化越大。
(2)前线轨道主要分布在芳环和吡咯环上,电子从HOMO轨道跃迁到LUMO属于π→π*跃迁。
(3)双羰基氰吡咯化合物分子在不同溶剂中的最大吸收波长及吸收强度随溶剂极性的增加而增大。
参考文献:
[1] 成贞辉. 多取代多吡咯的合成与表征[D]. 保定: 河北大学, 2006: 59.
Chenzhen Hui. Synthesize and Characterized of Polysubstituted pyrroles[D]. Baoding: Heibei University, 2006: 59.
[2] 江润生, 李勇军, 高娟, 等. 基于吡咯并吡咯二酮的D-A化合物的合成及其光学性质研究[J]. 中国科学: 化学, 2016(10): 1131-1140.
Runsheng Jiang, Yongjun Li, Juan Gao, et al. Synthesis, optical and redox properties of donor-acceptor molecules based on diketopyrrolopyrrole[J]. Scientia Sinica Chimica, 2016(10): 1131-1140.
[3] 瞿祎. 基于吡咯并吡咯二酮和喹吖啶酮的荧光化学传感器[D]. 上海: 华东理工大学, 2012: 198.
Qu Wei. Fluorescent Sensors Based on Diketopyrrolopyrrole and Quinacridone Derivatives[D]. Shanghai: East China University of Science and Technology, 2012: 198.
[4] 邱斌. 基于异腈的有机荧光小分子的合成、荧光性质的研究及Hetero-Pauson-Khand反应的初探[D]. 青岛: 青岛科技大学, 2015: 90.
Qiu Bin. Construction and Study of Novel Small Organic Fluorescent Molecules and Primary Exploration of Heter-Pauson-Khand Reaction Based on Isocyanide[D]. Qingdao: Qingdao University of Science and Technology, 2015: 90.
[5] 夏树伟, 沙鹏艳, 于良民, 等. 8-羟基喹啉锰配合物电子结构和光谱性质的含时密度泛函研究[J]. 高等学校化学学报, 2008(6): 1234-1238.
Xia Shuwei, Sha Pengyan, Yu Liangmin, et al. TD-DFT study on electronic structure and spectrum properties of 8-hydroxyquinolinato manganese complex[J]. Chemical Journal of Chinese Universities, 2008(6): 1234-1238.
[6] Shan M, Liu Y, Xia S, et al. A strategy of integrating ultraviolet absorption and crosslinking in a single molecule: DFT calculation and experimental[J]. Journal of Molecular Structure, 2016, 1107: 249-253.
[7] 王加亮, 李晴, 阚玉和. ABAB型酞菁衍生物取代基对结构和电子光谱的影响[J]. 淮阴师范学院学报(自然科学版), 2016(2): 126-131.
Wang Jialiang, Li Qing, Kan Yuhe. The effects of different substituents on the structure and electronic spectrum of ABAB-type phthalocyanine[J]. Journal of Huaiyin Teachers College (Natural Science Edition), 2016(2): 126-131.
[8] 任忠海, 王冬梅, 丁伟璐, 等. DFT研究给/吸电子取代基对D-π-A型苯并噻唑衍生物光电性质及非线性光学性质的影响[J]. 原子与分子物理学报, 2016, 33(2): 201-208.
Ren Zhonghai, Wang Dongmei, Ding Weilu, et al. Influence of donor /acceptor substitution on photoelectric properties and nonlinear optical properties of D-π-A benzothiazole derivatives: a DFT study[J]. Journal of Atomic and Molecular Physics, 2016, 33(2): 201-208.
[9] 胡薛, 孙铭骏, 张向飞, 等. 褐煤分子片段结构电子吸收光谱的理论研究[J]. 高等学校化学学报, 2015(11): 2292-2296.
Hu Xue, Sun Mingjun, Zhang Xiangfei, et al. Theoretical study of electronic spectra of lignite structural units[J]. Chemical Journal of Chinese Universities, 2015(11): 2292-2296.
[10] 杨芳, 王海峰, 李权. 两种薁磺酰胺席夫碱衍生物分子的光谱和热力学性质计算研究[J]. 原子与分子物理学报, 2016(4): 575-580.
Yang Fang, Wang Haifeng, Li Quan. Computational study of molecular spectra and thermodynamic properties of two kinds of azulene sulfonamide schiff base derivatives[J]. Journal of Atomic and Molecular Physics, 2016(4): 575-580.
[11] 李洁琼, 李永红. 用密度泛函理论研究两种金属镍席夫碱配合物的电子结构和光谱性质[J]. 化学研究, 2014(6): 616-621.
Li Jieqiong, Li Yonghong. Study on the electronic structure and spectral properties of two Ni(Ⅱ)-Schiff base complexes by density functional theory[J]. Chemical Research, 2014(6): 616-621.
[12] Frisch M J E A, Trucks G W, Schlegel H B, et al. Gaussian 09, Revision A. 02[M]. CON: Gaussian Inc. Wallingford, 2009: 200.
[13] 林树昌, 曾泳淮. 化学分析[M]. 北京: 高等教育出版社, 1998.
Lin Shuchang, Zeng Yonghuai. Chemical Analysis[M]. Beijing: Higher Education Press, 1998.
[14] 石文艳, 吕志敏, 雷武 等. 两种荧光染料的合成及其量子化学研究[J]. 南京理工大学学报, 2011(6): 867-872.
Shi Wenyan, Lv Zhimin, Lei Wei, et al. Synthesis and quantum chemistry study of two fluorescent dyes[J]. Journal of Nanjing University of Science and Technology, 2011(6): 867-872.
[15] Karpicz R, Puzinas S, Sulskus J et al. Electronic properties of carbazole-fluorene-benzothiadiazole compounds revealed by time resolved spectroscopy and quantum chemistry calculations[J]. Chemical Physics, 2012, 404: 82-87.
