沙棘H3K9乙酰化修饰全基因组分析
2018-04-08高国日陈道国何彩云
高国日,张 彤,陈道国,何彩云,2*
(1 中国林业科学研究院林业研究所 国家林业局林木培育重点实验室,北京 100091;2 南京林业大学南方现代林业协同创新中心,南京 210037)
沙棘(Hippophaerhamnoides)是一种多用途、耐寒和耐旱的落叶灌木植物,也是一种理想的防治水土流失的树种,能够有效改良荒漠化土地,增加野生动物栖息地和保护农庄等[1]。沙棘的果肉可以被制成沙棘汁,是一种具有很高营养价值的饮料,含有丰富的维生素C~E和胡萝卜素等生物活性成分。沙棘油提取于沙棘的种子,富含不饱和脂肪酸[2]。沙棘果实中丰富的营养物质能够调节人体免疫力、保护肝脏、抗辐射、预防动脉硬化、抗肿瘤以及促进组织再生等。因其生态价值、药用价值和经济价值,沙棘被认为是一种重要的植物[3]。近年来,对沙棘分子生物学的研究,不仅获得了可用于沙棘基因组分析、分子繁殖和群体遗传分析的大量简单重复序列(SSR)[4]及表达序列标签(EST)[5],还获得了响应冷冻[6-7]和干旱胁迫[8]的大量基因和蛋白质表达[9]信息,确定了其在响应低温和干旱中的生物学功能,在沙棘发育不同时期代谢组和蛋白组联合分析中,深入了解了沙棘果实独特营养成分形成机理[10-11]。虽然不同沙棘果色的lncRNA研究[12]为研究表观遗传对沙棘基因的表达调控提供了思路,但是表观遗传学中组蛋白修饰因其修饰类型和位点的复杂性,对基因表达的调控同样具有重要的作用[13]。许多研究已经表明组蛋白乙酰化修饰广泛参与植物的生长发育过程[14]及应对外界环境变化[15]。染色质免疫共沉淀技术(ChIP)是研究生物体内蛋白质与DNA相互作用的有效方法,通常用于组蛋白修饰类型和转录因子结合位点的研究,与二代测序相结合的ChIP-seq技术,能够高效准确的在全基因组范围内检测组蛋白修饰与转录因子所调控的DNA区域[16-17]。当前对拟南芥、水稻等组蛋白修饰和转录因子所调控的基因进行了大量的研究,其中组蛋白H3K9ac修饰在植物逆境胁迫及生长周期中,具有重要的作用[18]。并且由于组蛋白的保守性和调控基因的广泛性,非常适合推广至其他物种的表观遗传研究,2014年Li等[19]优化了研究毛果杨表观遗传的试验过程,阐明了木质部形成与组蛋白修饰的关系。
虽然研究沙棘组蛋白修饰对沙棘基因的表达调控方式,能够阐明沙棘在生长周期和逆境胁迫下基因表达的调控机制[20],但是使用商业抗体研究沙棘组蛋白修饰尚未见报道。本研究通过蛋白质印迹法(Western blot)验证H3K9ac抗体与沙棘组蛋白的结合能力,并使用抗体进行后续的ChIP试验。对富集到的沙棘DNA片段进行高通量测序,从而绘制沙棘H3K9乙酰化修饰图谱并鉴定出沙棘H3K9乙酰化修饰调控基因,为后续研究组蛋白修饰对沙棘基因表达的调控方式奠定基础。
1 材料和方法
1.1 试验材料
本试验使用的材料为中国沙棘(Hippophaerhamnoidessubsp.sinensis),采自中国林业科学研究院科研温室大棚,生长条件为自然光照,昼/夜温度为20~30 ℃/10~15 ℃,相对湿度在60%~70%。所使用的H3K9ac(ab10812)抗体购买于美国Abcam公司。
1.2 试验方法
1.2.1染色质免疫共沉淀技术DNA片段富集和蛋白质印迹法检测取沙棘叶片2 g,剪碎并浸没在甲醛交联缓冲液中,抽真空处理。加入甘氨酸后,终止交联,取出样品,洗涤并吸干表明水分。液氮研磨至粉末状,多次抽提、过滤去除杂质,最终重新悬浮。预留出部分样品后,加入核酸裂解缓冲液,置冰上裂解。对交联成的DNA-蛋白质复合物超声波片段化。取一部分进行Western blot试验,具体步骤为:配制分离胶和浓缩胶,加样前将梳子拔出,每个点样孔上样不超过7.5 μL。浓缩胶在80 V,分离胶在160 V下电泳,在电泳过程中裁剪PVDF膜并进行处理。电泳结束,撬去玻璃板,将浓缩胶轻轻刮去,将分离胶卸下。将分离胶铺在电转架上,再铺上浸好的PVDF膜,装好板,加入电转液和冰块,100 V转膜80 min。电转后的膜用1×TBST漂洗5 min,置于封闭液中,4 ℃过夜。分别加入一抗(H3K9ac),用封闭液稀释,室温震荡混匀2 h。1×TBST洗膜4×5 min,孵二抗(HRP-羊抗兔),室温摇床孵育2 h。用1×TBST洗膜3×10 min,用吸纸吸去多余水分,将ELC显色反应液加在膜上,使反应液分布均匀后,吸去边缘多余反应液,放到显色器中,观察显色效果并拍照保存。
取另一部分进行解交联纯化及琼脂糖凝胶电泳检测。加入目的蛋白抗体,形成抗体-组蛋白-DNA复合物,利用Protein A beads沉淀该复合物,特异性富集与目的组蛋白结合的DNA片段,多次洗涤,去除非特异性DNA片段,纯化复合体,解交联,利用DNA纯化试剂盒纯化DNA片段[21,22]。用Qubit检测DNA片段的浓度,并保存于-80 ℃。
1.2.2高通量测序及数据分析将ChIP试验所得DNA片段利用Ovation Ultralow Library kit (NuGEN, Part No. 0330) 建立ChIP文库,然后使用HiSeq 2000(Illumina)对DNA片段进行测序。通过trim方式对原始数据的质量进行检测,去掉低质量的数据,获得clean reads 用于后续分析。利用Burrows Wheeler Aligner(BWA) 将样本的clean reads 匹配到参考基因组上以及进行定位质量分析和mapping效率的分析。将mapping后唯一匹配的unique reads 通过MACS2(q-value≤0.05)[23]完成峰检分析,预测富集区域的峰即为组蛋白修饰的区域。然后与基因组上基因的位置进行重叠分析,包含有峰的基因就确定为该基因具有组蛋白修饰。通过GOseq R数据包和KOBAS软件对基因进行GO功能分析和KEGG通路分析。
2 结果和分析
2.1 DNA片段化、H3K9ac抗体与复合物的结合特性以及片段富集
将交联后的DNA片段化,琼脂糖凝胶电泳检测结果如图1所示,3个沙棘片段化DNA与片段化前各样本主带明显,片段化DNA明显分布于100~500 bp之间,效果较好,可进行下一步纯化、富集实验。经Western blot检测, H3K9ac抗体与复合物的结合特性如图2所示,该抗体与复合物结合能力较强,在15~17kDa处有明显的阳性条带出现。用H3K9ac抗体对沙棘片段化DNA进行富集,解交联,最终获得的DNA浓度为0.845 ng/μL,是阳性对照input DNA浓度的13.63%(表1)。阴性对照IgG富集的DNA浓度太低,没有检测到数值(表1)。
2.2 沙棘H3K9ac修饰的全基因组分析
2.2.1ChIP-seq测序数据及基因组分布将构建的IP和Input文库进行高通量测序,最终分别得到6G和10G的数据量,分别包含约2.2×107条和3.6×107条原始序列(图3)。将原始序列经过质量检测去除低质量序列后得到分析序列,其中分析序列占原始序列的99%以上。将分析序列与自测沙棘参考基因组进行比对,2个样本分析序列定位到基因组上序列的百分比均在35%以上。唯一定位的测序序列占定位到基因组上序列的80%左右。对唯一比对序列比对到基因组的各条染色体上的密度进行统计,结果如图4所示,序列在各条染色体上均有分布,并且在染色体上的不同区域富集程度不同。序列相对于基因位置分布表明序列在结构基因中的分布要高于基因上下游部分,并且在结构基因中的两端具有明显的富集(图5)。
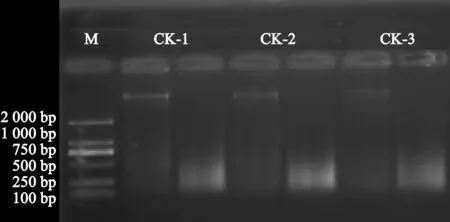
M. DNA分子标量;CK-1、CK-2、CK-3.3个生物学重复图1 沙棘DNA片段化结果M.DNA Marker;CK-1,CK-2,CK-3.Three biological repeats Fig.1 The results of DNA fragment of H. rhamnoides
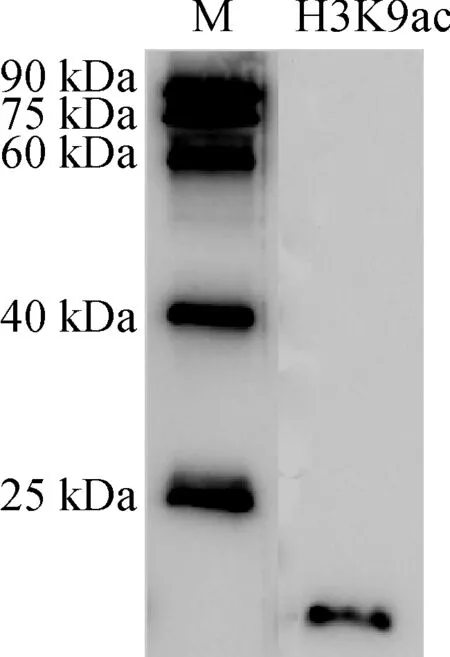
图2 H3K9ac与复合物结合特性Fig.2 The binding specificity of histone with H3K9ac antibody

样品名称Samplename试验用量Dosage/μL浓度Content/(ng/μL)溶解体积Volume/μLInputCK206.2012IPCK-1(H3K9ac)200.8914IPCK-2(H3K9ac)200.8014IPCK-3(IgG)20low13
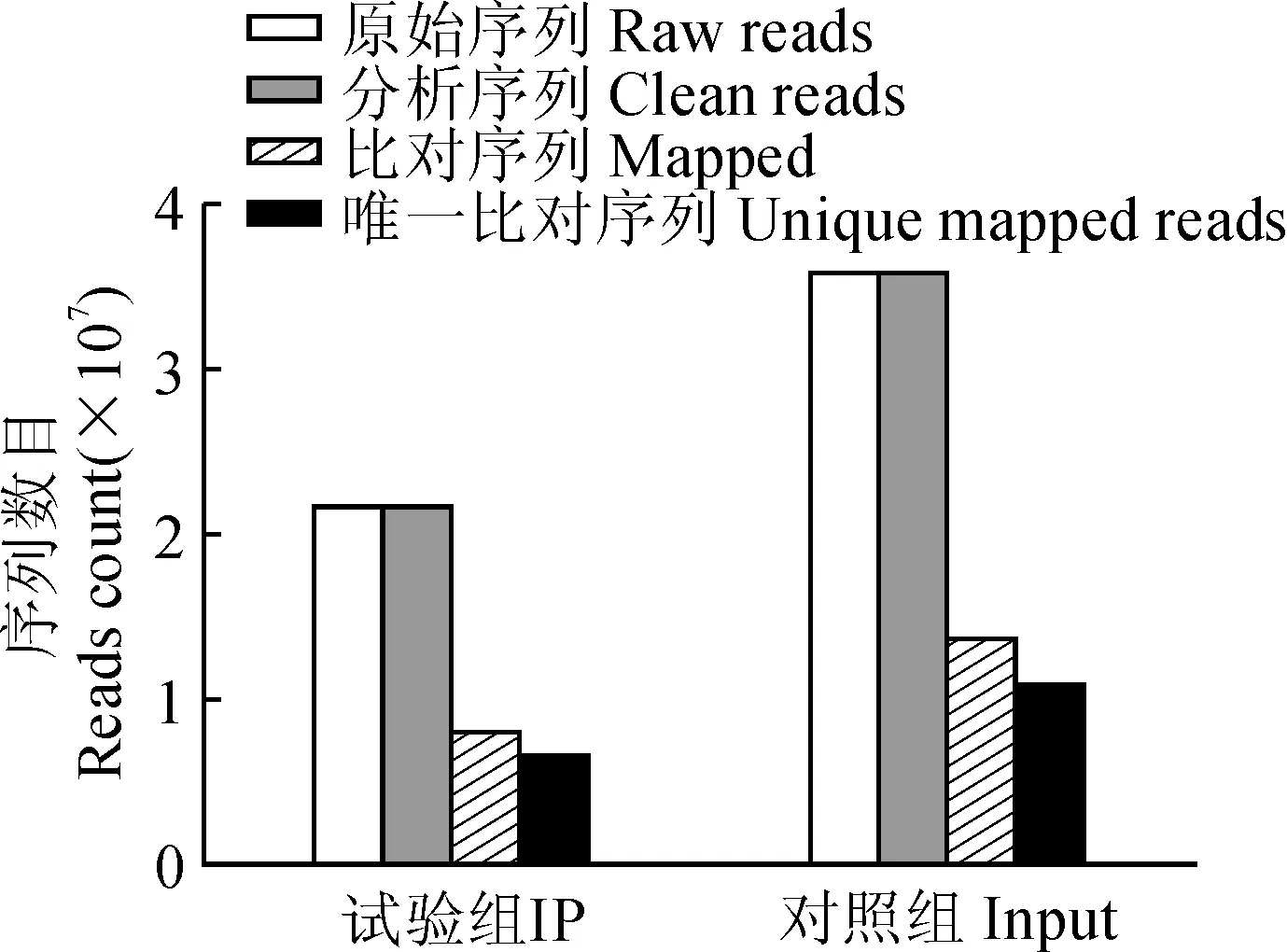
图3 测序数据及与参考基因组序列的比对统计Fig.3 The statistics of sequencing data and mapping with reference genome
2.2.2富集区域峰的预测及基因注释对mapping到参考基因组上的unique reads的富集区进行峰(peak)的预测,每个peak代表一个H3K9ac的修饰位点。共预测出1 011个peak,长度为147 bp,peak中所包含的reads的个数占所有比对上的reads的20.83%。Peak在染色体上的分布如图6所示。从图6可以看出,H3K9ac修饰位点在不同染色体上均匀分布。对修饰基因进行GO功能注释,包含基因数最多的前30类结果如图7所示,H3K9ac修饰可以调控的基因功能包括了生物学过程(17类)、分子功能(5类)和细胞组成(8类),其中,在生物学过程分类中,包含基因数最多的是细胞生理过程和细胞生长中代谢过程,在细胞组成中,膜质和细胞分类中拥有的基因数最多,分子功能中结合作用占有最大比例。KEGG通路分析结果表明,基因在50个代谢通路中得到注释,包含基因数最多的主要有:代谢途径、氧化磷酸化和次级代谢物的合成。这些过程为研究H3K9ac修饰所调控基因的功能提供了新的信息。
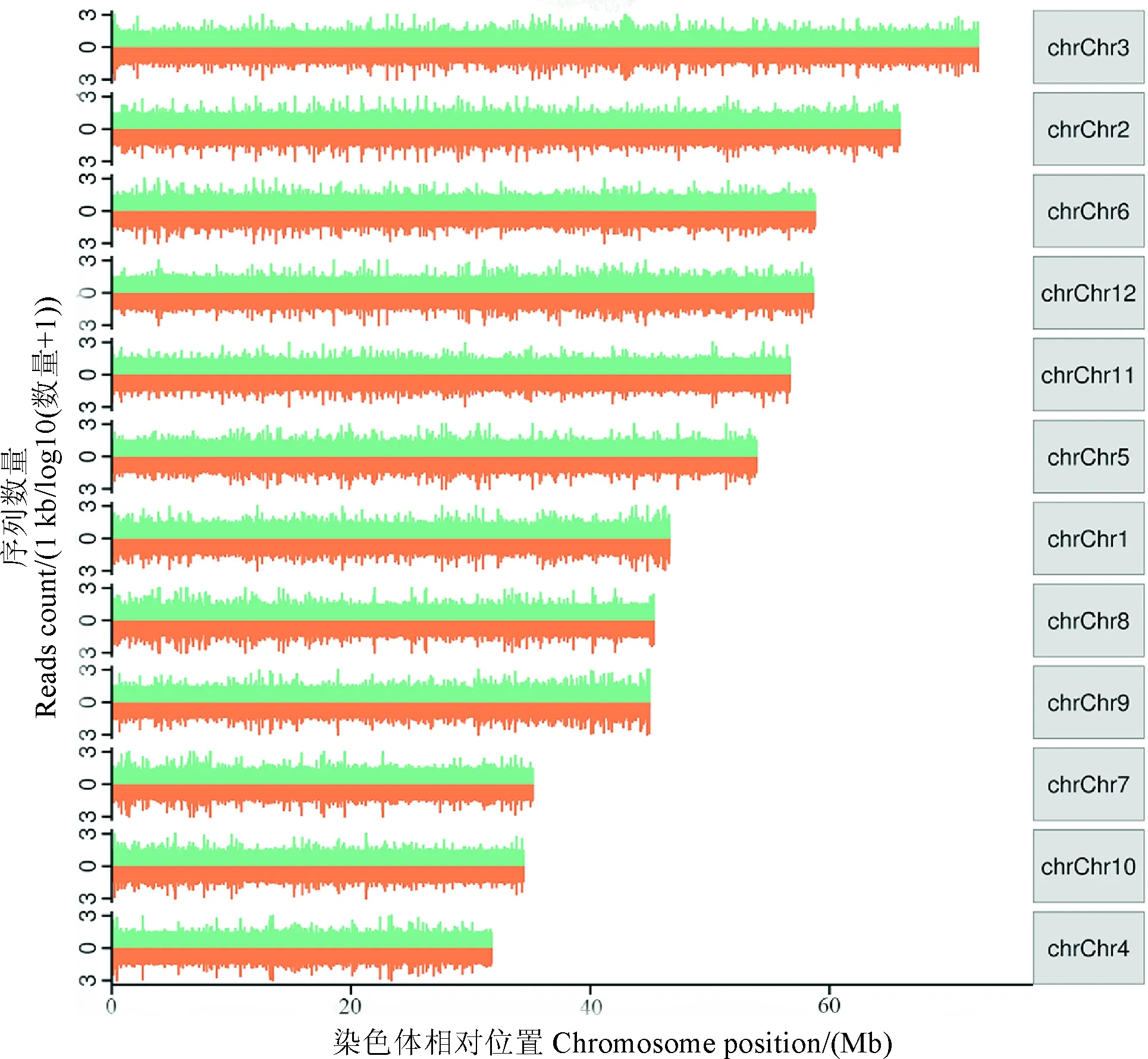
图4 IP的reads在基因组上的分布Fig.4 The distribution of reads of IP in genome

图5 IP的reads相对基因位置的分布Fig.5 The distribution of reads of IP in relative genetic location

图6 Peak在染色体上的分布Fig.6 The distribution of peak on chromosome
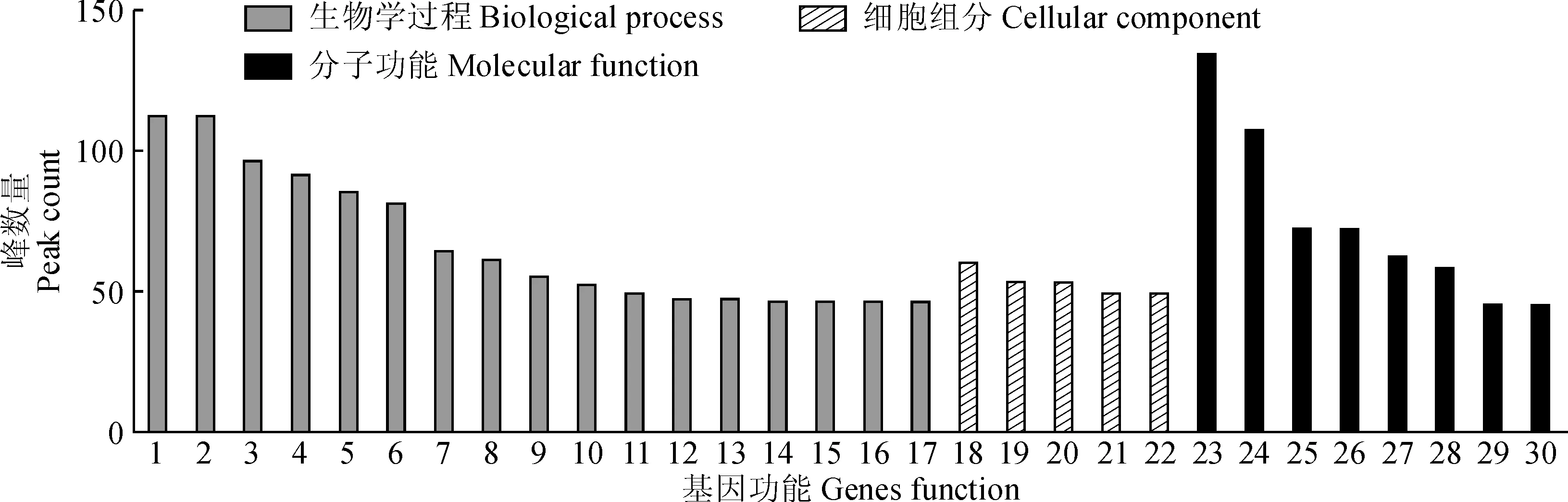
1.细胞过程;2.代谢过程;3.有机物代谢过程;4.初级代谢过程;5.单个组织过程;6.细胞代谢过程;7.单个组织细胞过程;8.高分子代谢过程;9.细胞高分子代谢过程;10.氮化合物代谢过程;11.细胞氮化合物代谢过程;12.单生物代谢过程;13.生物合成过程;14.杂环代谢过程;15.细胞生物合成过程;16.有机环状化合物合成代谢过程;17.有机物生物合成过程;18.细胞膜;19.细胞;20.细胞部分;21.胞内部分;22.胞内;23.结合;24.催化活性;25.有机环状化合物结合;26.杂环化合物结合;27.离子结合;28.蛋白结合;29.核酸结合;30.水解酶活性图7 GO功能富集分布图1.Cellular process;2.Metabolic process;3.Organic substance metabolic process;4.Primary metabolic process;5.Single-organism process;6.Cellular metabolic process;7.Single-organism cellular process;8.Macromolecule metabolic process;9.Cellular macromolecule metabolic process;10.Nitrogen compound metabolic process;11.Cellular nitrogen compound metabolic process;12.Single-organism metabolic process;13.Biosynthetic process;14.Heterocycle metabolic process;15.Cellular biosynthetic process;16.Organic cyclic compound metabolic process;17.Organic substance biosynthetic process;18.Membrane;19.Cell;20.Cell part;21.Intracellular part;22.Intracellular;23.Binding;24.Catalytic activity;25.Organic cyclic compound binding;26.Heterocyclic compound binding;27.Ion binding;28.Protein binding;29.Nucleic acid binding;30.Hydrolase activityFig.7 The distribution of GO function

通路名称Pathwayname基因数Genenumber通路名称Pathwayname基因数Genenumber新陈代谢通路Metabolicpathways23核黄素代谢Riboflavinmetabolism1氧化磷酸化Oxidativephosphorylation9丁酸甲酯代谢Butanoatemetabolism1次生代谢物合成Biosynthesisofsecondarymetabo-lites9牛磺酸和亚牛磺酸代谢Taurineandhypotaurinemetabolism 1RNA转运RNAtransport4错配修复Mismatchrepair1淀粉和蔗糖代谢Starchandsucrosemetabolism4玉米素合成Zeatinbiosynthesis1果糖和甘露糖代谢Fructoseandmannosemetabo-lism3缬氨酸、亮氨酸和异亮氨酸生物合成Valine,leu-cineandisoleucinebiosynthesis1核苷酸切除修复Nucleotideexcisionrepair3丙胺酸代谢beta-Alaninemetabolism1丙酮酸代谢Pyruvatemetabolism3维生素C代谢Ascorbateandaldaratemetabolism1糖酵解/糖异生途径Glycolysis/Gluconeogenesis3碱基切除修复Baseexcisionrepair1氨基糖和核苷酸糖代谢Aminosugarandnucleo-tidesugarmetabolism3丙三酸、天冬氨酸和谷氨酸代谢Alanine,aspartateandglutamatemetabolism1胞吞作用Endocytosis3DNA复制DNAreplication1内质网蛋白加工Proteinprocessinginendoplasmicreticulum 3二羧酸代谢Glyoxylateanddicarboxylatemetabo-lism1碳代谢Carbonmetabolism3蛋白质运输Proteinexport1磷酸戊糖途径Pentosephosphatepathway2甘油酯代谢Glycerolipidmetabolism1半乳糖代谢Galactosemetabolism2核糖体Ribosome1同源重组Homologousrecombination22-氧代羧酸代谢2-Oxocarboxylicacidmetabolism1蛋白酶体Proteasome2精氨酸和脯氨酸代谢Arginineandprolinemetabo-lism1光和生物的碳固定Carbonfixationinphotosyn-theticorganisms2戊糖和葡萄糖醛酸转换Pentoseandglucuronateinterconversions 1光合作用Photosynthesis2半胱氨酸和甲硫氨酸代谢Cysteineandmethioninemetabolism 1谷胱甘肽代谢Glutathionemetabolism2植物病原菌互作Plant-pathogeninteraction1RNA降解RNAdegradation2嘧啶代谢Pyrimidinemetabolism1mRNA监测途径mRNAsurveillancepathway2嘌呤代谢Purinemetabolism1泛素介导的蛋白水解作用Ubiquitinmediatedpro-teolysis2真核细胞核小体合成Ribosomebiogenesisineu-karyotes1氨基酸生物合成Biosynthesisofaminoacids2剪接体Spliceosome1植物信号激素转导Planthormonesignaltransduc-tion2甘氨酸、丝氨酸和苏氨酸代谢Glycine,serineandthreoninemetabolism1
3 讨 论
动植物在不同的进化中,组蛋白的氨基酸序列非常保守,即使存在较远亲缘关系,4种组蛋白(H2A、H2B、H3和H4)氨基酸序列都非常相似[24]。所以,可以使用同一种抗体研究不同物种的组蛋白修饰。但是,不同物种间的组蛋白还是存在微小的空间结构差异,所以同一抗体对不同物种组蛋白的结合能力会有所差别[25]。对沙棘染色质进行超声波破碎后,片段大小集中在100~500 bp,说明该试验条件的适用性。通过Western blot检测发现H3K9ac抗体与沙棘片段化染色质具有较强的结合能力,可以用于后续的富集试验。用H3K9ac抗体对沙棘片段化DNA进行富集,最终富集得到的DNA浓度为0.845 ng/μL,仅为阳性对照Input DNA浓度的13.63%,低于对拟南芥[21]、毛果杨所报道的浓度[19],原因可能是抗体与沙棘组蛋白的结合能力有限。通过阴性对照IgG可以确定抗体与组蛋白的特异性良好。DNA片段富集浓度低的原因可能是试验过程中的试验条件并不是最优方案,所选用抗体存在物种差异,或者是沙棘中H3K9ac修饰密度低于拟南芥和毛果杨等,可以通过改进试验方案或定制抗体来提高富集浓度,进一步进行验证。
ChIP-seq获得的数据,在经过一系列的质量检测和筛选后,clean reads将被用于后续的分析。由于ChIP获得的是DNA片段,其所测定的序列与参考的基因组的mapping率应该在70%以上[26-27],但是,在本研究中2个样本的mapping率只有35%,造成该现象的原因可能是因为自测基因组中含有大量的重复序列,确切的结论还需要在进一步完善沙棘基因组后才能获得。Reads在染色体上分布广泛,更加证明了组蛋白H3K9ac修饰对于沙棘基因的表达具有广泛的调控作用[28],reads相对基因位置的分析表明,H3K9ac对于沙棘基因的调控主要发生在结构基因的两端。由于染色体被超声波随机打断,所以在通过特异抗体富集后,片段之间的存在大量的重叠区域,为了更加准确分析基因功能,将reads富集区域称为peak,即为DNA与相应组蛋白的结合位点。我们利用GO数据库和KEGG数据库,对peak所在区域的基因进行GO功能富集和KEGG通路分析,不仅可以预测基因功能,还可以研究基因在不同代谢通路中的位置和作用,通过注释,准确获得了H3K9ac修饰所能够调节的基因功能,主要涉及到生物学过程中的细胞生理过程和细胞生长中代谢过程,细胞组成中的膜质和细胞分类,以及分子功能中小分子间有选择性、非共价结合作用。在代谢途径、氧化磷酸化和次级代谢物的合成通路中具有重要的作用。综上所述,H3K9ac调控基因对于沙棘基因的正常表达具有重要的作用。
本研究通过western blot验证了H3K9ac抗体与复合物具有较强的结合能力。沙棘片段化DNA的富集以及高通量测序证明了抗体能够用于研究沙棘的组蛋白修饰类型,并且绘制了沙棘第一张H3K9ac修饰遗传图谱草图和鉴定出沙棘H3K9ac修饰所调控基因,为今后研究组蛋白修饰对沙棘基因表达的调控方式奠定基础。通过对试验条件的优化,提高抗体对组蛋白的捕获效率,将会获得更加完整的组蛋白修饰遗传图谱。
参考文献:
[1]LI T S, SCHROEDER W R. Sea buckthorn (HippophaerhamnoidesL.): a multipurpose plant[J].Horttechnology, 1996,6(4):370-380.
[2]BEVERIDGE T, LI T S, OOMAH B D,etal. Sea buckthorn products: manufacture and composition[J].JournalofAgricultural&FoodChemistry, 1999,47(9):3 480-3 488.
[3]SURYAKUMAR G, GUPTA A. Medicinal and therapeutic potential of sea buckthorn (HippophaerhamnoidesL.)[J].JournalofEthnopharmacology, 2011,138(2):268-278.
[4]JAIN A, GHANGAL R, GROVER A,etal. Development of EST-based new SSR markers in sea buckthorn[J].PhysiologyandMolecularBiologyofPlants, 2010,16(4):375-378.
[5]JAIN A, CHAUDHARY S, SHARMA P C. Mining of microsatellites using next generation sequencing of sea buckthorn (HippophaerhamnoidesL.) transcriptome[J].PhysiologyandMolecularBiologyofPlants, 2014,20(1):115-123.
[6]GHANGAL R, RAGHUVANSHI S, SHARMA P C. Expressed sequence tag based identification and expression analysis of some cold inducible elements in sea buckthorn (HippophaerhamnoidesL.)[J].PlantPhysiology&Biochemistry, 2012,51(2):123-128.
[7]GHANGAL R, SHARMA P C. DeepSAGE based differential gene expression analysis under cold and freeze stress in sea buckthorn (HippophaerhamnoidesL.)[J].PloSOne, 2015,10(3):e0121982.
[8]HE C, ZHANG G, ZHANG J ,etal.Phosiological, biochemical, and proteome profiling reveals key pathways underlying the drought stress responses ofHippophaerhamnoides[J].Proteomics, 2016,16(20):2 688-2 697.
[9]HE C, GAO G, ZHANG J,eral. Proteome profiling reveals insights into cold-tolerant growth in sea buckthorn[J].ProteomeScience,2016,14(1):14.
[10]FATIMA T, SNYDER C L, SCHROEDER W R,etal. Fatty acid composition of developing sea buckthorn (HippophaerhamnoidesL.) berry and the transcriptome of the mature seed[J].PloSOne,2012,7(4):e34099.
[11]HE C, ZHANG G, ZHANG J,etal. Integrated analysis of multiomic data reveals the role of the antioxidant network in the quality of sea buckthorn berry[J].FasebJournal, 2017,31(5):1 929-1 938.
[12]ZHANG G, DUAN A, ZHANG J,etal. Genome-wide analysis of long non-coding RNAs at the mature stage of sea buckthorn (HippophaerhamnoidesLinn) fruit[J].Gene, 2017, 596:130-136.
[13]GRANT-DOWNTON R T, DICKINSON H G. Epigenetics and its implications for plant biology. 1. the epigenetic network in plants[J].AnnalsofBotany, 2005,96(7):1 143-1 164.
[14]KOMET N, SCHERES B. Members of the GCN5 histone acetyltr ansferase comples regulate PLETHOR-mediated root stem cell niche maintenance and transit amplifying cell proliferation inArabidopsis[J].PlantCell,2009,21(4):1 070-1 079.
[15]LUO M, WANG Y, LIU X,etal.HD2C interacts with HDA6 and is involved in ABA and salt stress response inArabidopsis[J].JournalofExperimentalBotany, 2012,63(8):3 297-3 306.
[16]SCHMIDT D, WILSON M D, SPYROU C,etal. ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions[J].Methods, 2009,48(3):240-248.
[17]LANDT S G, MARINOV G K, KUNDAJE A,etal. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia[J].GenomeResearch, 2012,22(9):1 813-1 831.
[18]ASENSIFABADO M A, AMTMANN A, PERRELLA G. Plant responses to abiotic stress: The chromatin context of transcriptional regulation[J].BiochimicaetBiophysicaActa, 2016,1860(1):106-122
[19]LI W, LIN Y C, LI Q,etal. A robust chromatin immunoprecipitation protocol for studying transcription factor-DNA interactions and histone modifications in wood-forming tissue[J].NatureProtocols, 2014,9(9):2 180-2 193.
[20]潘丽娜,王振英.植物表观遗传修饰与病原菌胁迫应答研究进展[J].西北植物学报,2013,33(1): 210-214.
PAN L N, WNAG Z Y. Epigenetic control in plant pathogen-stress response[J].ActaBotanicaBoreali-OccidentaliaSinica,2013,33(1):210-214.
[21]SALEH A, ALVAREZ-VENEGAS R, AVRAMOVA Z. An efficient chromatin immunoprecipitation (ChIP) protocol for studying histone modifications inArabidopsisplants[J].NatureProtocols, 2008,3(6):1 018-1 025.
[22]KAUFMANN K, MUINO J M, ØSTERÅS M,etal. Chromatin immunoprecipitation (ChIP) of plant transcription factors followed by sequencing (ChIP-SEQ) or hybridization to whole genome arrays (ChIP-CHIP)[J].NatureProtocols, 2010,5(3):457.
[23]SALMON-DIVON M, DVINGE H, TAMMOJA K,etal. PeakAnalyzer: genome-wide annotation of chromatin binding and modification loci[J].BmcBioinformatics, 2010,11(1):415.
[24]DELANGE R J, SMITH E L. Histone function and evolution as viewed by sequence studies[C].New York:JohnWiley&Sons, Ltd, 2008:59-76.
[25]袁亚静,许静,王伟.Chromodomain蛋白在表观遗传调控中的功能[J].中国生物化学与分子生物学报,2016,(4):359-364.
YUAN Y J, XU J,WANG W.The role of chromodomain-containing proteins in epigenetic regulation[J].ChineseJournalofBiochemistryandMolecularBiology,2016,(4):359-364.
[26]ZHANG X, CLARENZ O, COKUS S,etal. Whole-genome analysis of histone H3 lysine 27 trimethylation inArabidopsis[J].PloSBiology, 2007,5(5):e129.
[27]ZHANG X, BEMATAVICHUTE Y V, COKUS S,etal. Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 inArabidopsisthaliana[J].GenomeBiology, 2009,10(6):1-14.
[28]王淑妍,郭九峰,刘晓婷,等.非生物胁迫下植物表观遗传变异的研究进展[J].西北植物学报,2016,36(3):631-640.
WANG S Y, GUO J F, LIU X T,etal. Research progress of abiotic stress induced epigenetic variation in plants.[J].ActaBotanicaBoreali-OccidentaliaSinica,2016,36(3):631-640.
