利用概率估算提高植物品种分子标记鉴定的准确率
2018-04-03周俊飞崔野韩唐浩李论陈红温雯韩瑞玺黄思思方治伟彭海
周俊飞,崔野韩,唐浩,李论,陈红,温雯,韩瑞玺,黄思思,方治伟,彭海
(1江汉大学系统生物学研究院,武汉 430056;2农业部科技发展中心,北京 100122)
0 引言
【研究意义】依据现有的标准进行植物品种分子鉴定是植物品种权授予、品种审定、种业市场管理与品种权案件判决的重要依据。然而,现有的品种分子鉴定标准仅检测了植物品种基因组上部分分子标记位点,存在抽样误差,但标准中并没有给出抽样误差的估算方法与鉴定结论的可靠性估算方法,导致鉴定结论在应用中常被质疑是否可靠。估算并验证分子标记位点的抽样误差与鉴定结论的可靠性,为品种分子鉴定标准的各种应用提供科学依据。【前人研究进展】植物品种鉴定分为田间表型测试与分子标记测试2种方法。田间种植鉴定通过观察申请品种与近似品种在相同生长条件下的性状差异,判断申请品种是否存在特异性。为保证鉴定结果的可靠性,田间试验通常需持续2—3年。田间表型测试具有周期长、受环境影响等缺点。分子标记测试则不受环境影响,可以快速检测品种间的异同,从而辅助实现品种快速授权或品种权侵权案件的判决。分子标记鉴定方法通过对分子标记位点进行检测,将申请品种与近似品种间的差异标记数与规定的阈值进行比较,判断两者是否为不同的品种。目前,品种分子标记鉴定主流技术为微卫星(simple sequence repeats,SSRs)标记。SSR标记是利用基因组内广泛存在的、中性的简单重复序列的长度变异鉴定品种,具有标记多态性高[1]、鉴定方法简单、种内[2-3]甚至种间[4-6]通用等突出优势。目前,大多数植物品种分子鉴定行业标准或者国家标准都是根据SSR标记制定的。然而,SSR标记具有检测通量低等不足,有逐步被单核苷酸变异(single nucleotide polymorphisms,SNPs)标记所取代的趋势,目前,部分作物如水稻[7]等已经基于 SNP标记制定了品种鉴定的行业标准。【本研究切入点】任何分子标记方法都只能鉴定品种基因组上的部分位点,必然存在标记位点的抽样误差。大多数基于SSR标记的品种分子鉴定标准的标记位点数不足50个,抽样误差十分明显。因而,利用现有的品种分子鉴定标准获得的鉴定结论用于品种权授权、品种审定和解决品种权纠纷时,可靠性常常被质疑。然而,现有的植物品种分子鉴定标准中均无标记位点误差与结论可靠性的计算方法,相关研究也十分有限。【拟解决的关键问题】本研究估算了品种分子标记鉴定的位点抽样误差与结论的可靠性,进而将品种间关系划分为红区、绿区和黄区,对应高概率保障为相同或近似品种、高概率保障为不同品种和无概率保障的待定品种,为品种分子鉴定的行业标准与国家标准的应用提供了科学依据。
1 材料与方法
1.1 品种间关系鉴定的概率模型
设品种分子鉴定的行业标准或国家标准中规定的检测位点的数目为N个,品种A和B间观察到的差异位点的数目为 x(x≤N)个。标准中规定区分相同和不同品种的差异位点的数目的阈值标准为 n(n≤N)个。那么,根据行业标准或国家标准,若x<n,则判定品种A和B为相似或相同品种,否则,为不同品种。
由于N个检测位点只是基因组上所有标记位点的一次抽样,因此,观察到的差异位点的数目x值的大小存在抽样误差。排除抽样误差后,品种A和B间的真实的差异位点(后文称为期望的差异位点)数目 t值可以更加准确地判定品种A和B的关系。根据分子鉴定标准,品种A与B为相同品种的概率为P=∑0≤t<nP(t|x),其中,P(t|x)为观察到的差异位点的数目为 x的条件下,期望的差异位点的数目为t的条件概率。根据贝叶斯公式:
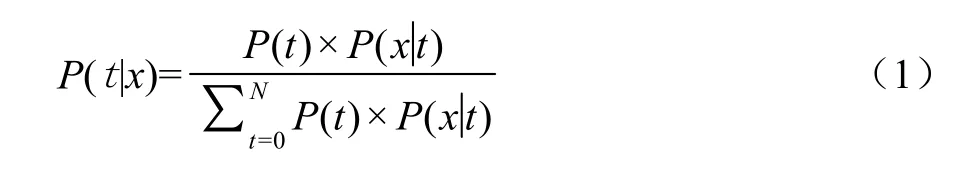
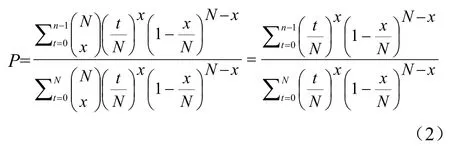
设接受品种A与B为相同品种的概率保障为1-α,那么,当P≥1-α,品种A与B的关系落入红区,判定它们为相同品种或近似品种;当P≤α时,品种A与B的关系落入绿区,判定它们为不同品种;当α<P<1-α时,品种A与B的关系落入黄区,无法准确判定它们之间的关系。
1.2 品种间关系鉴定的概率模型的验证
水稻品种分子鉴定标准[8]规定的检测位点为N=48个SSR标记位点,区分相同与不同品种的差异位点的数目,即判定阈值 n=2。为验证概率模型,共选择了8个水稻品种,利用AmpSeq-SSR的方法为每个品种鉴定了3 205个SSR标记位点,品种的具体名称见表1,SSR标记位点选取、检测方法与检测结果见我们近期发表的论文[9]。3 205个标记位点抽样量较大,抽样误差小,因此,品种间期望的差异位点的数目可以估计为其中,M(M≤3 205)为两品种中均成功获得了基因分型的检测位点的数目;m为共同检出位点中,观察到的差异位点的数目。根据水稻分子鉴定标准,当t<n=2时,两品种为相同或相似品种,否则为不同品种,该判定结果为品种间关系的参考值。
从2个品种共同检测位点中随机抽取N=48个检测位点,获得每次抽样中观察到的差异位点的数目,即x的值。根据公式(2)计算P值,并与设定的α值比较,判定品种间的关系,称为品种间关系的判定结论。将品种间关系的判定结论与参考值比较,验证判定结论是否正确。
2 结果
2.1 利用概率模型可以准确鉴定品种间关系
对8个水稻品种的28对组合的3 205个SSR标记的分型结果进行了10 000次N=48个标记位点的随机抽样,每一次随机抽样相当于利用水稻SSR标记鉴定的行业标准[8]对一对品种进行了一次鉴定。利用公式(2)所示的概率模型,根据每次随机抽样结果判定品种组合间的关系,并验证判定结论的正确性,具体过程见材料与方法部分。结果表明,每个品种组合有 4 295—10 000(42.95%—100%)次随机抽样在1-α=0.95的概率保障下,明确判定为不同品种(表1),品种间关系落入了绿区。把表1中的t值与水稻SSR标记鉴定的行业标准[8]中的判定阈值n=2比较,获得每一对品种组合间关系的参考值。品种组合间关系的参考值与判定结论比较表明,所有(100%)判定结论均是正确的,表明利用概率模型获得的品种间关系的判定结论的准确性很高。
2.2 概率模型在品种权案件判决中的应用实例
最近(2017年8月3号),根据西瓜品种‘美丰’与‘欣美’间观察到的差异位点的数目x=7个,合肥市中级人民法院判决它们之间不构成品种权侵权;根据‘美丰’与‘辉煌818’,‘美丰’与‘兴隆 188’之间观察到的差异位点的数目 x=0个,判决它们之间构成品种权侵权。但败诉方认为西瓜分子鉴定标准[10]只检测了 N=28个位点,质疑判决的可靠性。
西瓜分子鉴定标准中,相同与不同品种的判定阈值n=3,根据公式(2),当x=7时,为相同品种的概率P=2.49×10-3≤α=0.05。因此,‘美丰’与‘欣美’在95%的概率保障下落入了绿区,为不同品种,不构成侵权的判决是可靠的。当x=0时,P=2.96≥1-α=0.95,因此,‘美丰’与‘辉煌 818’,‘美丰’与‘兴隆188’在 95%的概率保障下落入了红区,为相同或相似品种,构成侵权判决也是可靠的,败诉方质疑的理由并不充分。
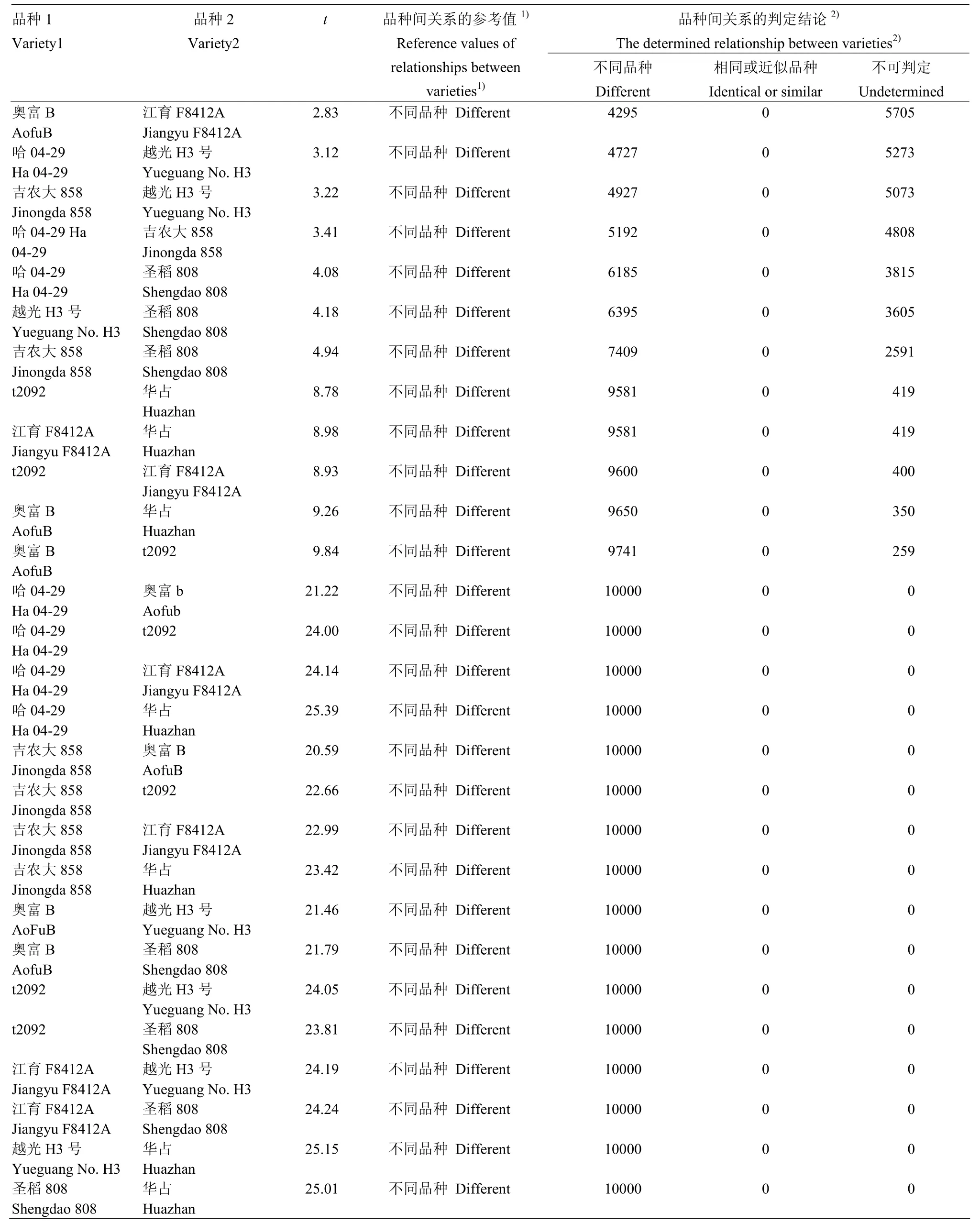
表1 利用概率模型可以准确判定品种间关系Table 1 The relationship between varieties can be determined by probability model
3 讨论
3.1 假设t值服从均匀分布的理由
差异大的品种可以根据性状直接判定为不同品种,也就是说,多数情况下,利用分子标准鉴定的品种差异不大。因此,期望的差异位点的数目t值接近于品种鉴定标准中区分相同与不同品种的判定阈值n,且t值与n值之间的差值大小接近随机事件,导致t值的分布服从于以n为平均值的平缓的正态分布,即t~N(n, σ2)。当在授权审查或案件判决中,获得多对性状间差异不大的品种间的 DNA分子标记差异数量的实测值后,就可以计算出 σ2的值和公式(1)中的先验概率 P(t)的值,最终精确计算出相同品种的后验概率P值并精确判定品种间的关系。
当品种间 DNA分子标记差异数量的实测值数据有限,导致不能或不能准确计算公式(1)中的先验概率P(t)的值时,可用t值的均匀分布近似估计正态分布,将公式(1)转化为公式(2),实现相同品种的后验概率P值的估算。由于t值的分布服从于以n为平均值的平缓的正态分布,因此,近似为均匀分布是合理的。根据相同品种概率P值的计算公式的原理,将正态分布近似为均匀分布,所产生的效应仅是略为增加判定品种间关系的黄区的范围,同时略为缩小红区与绿区的范围。也就是说,使用近似的均匀分布,不能明确获得品种间关系的机会略有增加,但获得的明确鉴定结论更加可靠,这符合品种授权与案件判决对品种鉴定结论的精准性的要求。
当品种间差异较大,即 t值与 n值之间的差值较大时,无论是采用均匀分布还是正态分布,为相同品种的后验概率P的值都很低,因此,均可正确地判定出它们为不同的品种。事实上,表1中差异大的品种间的关系判定全部得到了验证,表明均匀分布的假定基本不会影响差异大的品种间关系判定的准确率。
总之,假定t值服从均匀分布,实现了相同品种的后验概率P值计算的同时,对品种间关系判定结论的准确性也基本无影响。
3.2 现有品种分子鉴定标准中检测位点不足导致鉴定结论不明确
现有的大部分基于SSR标记的品种鉴定标准中规定的检测位点都较少,例如,水稻品种分子鉴定标准[8]规定检测位点数仅为N=48个。即使在品种间没有观察差异分子标记位点,即x=0,根据公式(2),它们为相同品种或相似品种的概率 P=0.87,落入了黄区,无法在1-α=0.95的概率保障下,判定为相同或近似品种。事实上,表1显示每个品种组合中有0—5 705(0—57.05%)次抽样,不能在 1-α=0.95的概率保障下得到品种组合间关系的明确结论,品种组合间关系落入了黄区,需要结合其他证据如田间测试结果进行判定。
增加品种分子鉴定标准中检测位点的数目可解决品种间关系鉴定结论不明确的问题。假设将水稻品种分子鉴定标准[8]中检测位点数量和判定阈值同时扩大10倍(N=480且n=20),那么,判定相同与不同品种间的分子差异水平并没有改变。若在品种间依然没有观察到差异标记位点(x=0),那么根据公式(2),它们为相同或相似品种的概率 P=1.00。也就是说,几乎有绝对的把握保证这品种间的分子差异在标准规定的判定阈值之下,可以明确地判定为相同或相似的品种。
总之,增加分子鉴定标准中检测位点的数量,可以更精准地评估品种间分子差异水平,增加具有明确判定结论的红区与绿区的范围,缩小不具有明确判定结论的黄区的范围。
3.3 分子标记鉴定在品种鉴定中的作用与地位
品种分子鉴定存在以下局限。首先,分子鉴定标准只抽检基因组上部分位点,存在抽样误差,不能准确说明品种间的分子差异水平。本研究为品种分子鉴定结论赋予了概率保证,减少了因检测位点不足导致的争议。第二,分子标记分型技术无法做到绝对准确,导致品种间分子差异的假阳性与假阴性。例如,SNP分型的技术误差超过1%[11-12],聚合酶滑动[13-19]也造成大量 SSR标记分型错误[9]。最近发明的AmpSeq-SSR技术有效解决了SSR标记检测位点不足与分型错误的问题[9]。第三,碱基突变率约为10-9/世代[20],而多数作物基因组超过109个碱基,导致相同品种间也可能存在约2个标记的差异。因此,存在差异的分子标记不能直接作为不同品种的判定依据。
品种鉴定是品种权授权与案件判决的依据,需同时满足科学与法律的严谨性,需要逻辑完整的解决方案。品种分子标记鉴定弥补了性状鉴定速度慢的不足,但《种子法》是基于性状定义品种的,决定了分子标记鉴定不具有最终的法律地位。品种间分子标记差异位点的多少反应了它们为相同或不同品种概率的大小,可以依据概率大小快速作出预授权或判决,以充分利用分子标记鉴定速度快的优势。然而,类似于刑事案件,分子标记鉴定结果的孤证不足以成为最终定案依据。当预授权或判决存在争议时,可根据分子鉴定结果确定举证责任方,举证责任方可以根据性状测试结果,论证请求的合理性。
4 结论
构建了一个评估品种间关系判定结论可靠性的概率模型,利用该概率模型鉴定的品种间关系的准确率为 100%。为品种间关系的分子鉴定结论赋予了概率保证,提高品种间关系判定结论的准确性,避免因检测位点不足导致的争议。
[1]BUSCHIAZZO E, GEMMELL N J. The rise, fall and renaissance of microsatellites in eukaryotic genomes. Bioessays, 2006, 28(10):1040-1050.
[2]MOORE S, SARGEANT L, KING T, MATTICK J, GEORGES M,HETZEL D. The conservation of dinucleotide microsatellites among mammalian genomes allows the use of heterologous PCR primer pairs in closely related species. Genomics, 1991, 10(3): 654-660.
[3]MOODLEY Y, BAUMGARTEN I, HARLEY E. Horse microsatellites and their amenability to comparative equid genetics. Animal Genetics,2006, 37(3): 258-261.
[4]DAWSON D A, HORSBURGH G J, KÜPPER C, STEWART I R,BALL A D, DURRANT K L, HANSSON B, BACON I, BIRD S,KLEIN A. New methods to identify conserved microsatellite loci and develop primer sets of high cross species utility-as demonstrated for birds. Molecular Ecology Resources, 2010, 10(3): 475-494.
[5]MOODLEY Y, MASELLO J F, COLE T L, CALDERON L,MUNIMANDA G K, THALI M R, ALDERMAN R, CUTHBERT R J,MARIN M, MASSARO M, NAVARRO J, PHILLIPS R A, RYAN P G,SUAZO C G, CHEREL Y, WEIMERSKIRCH H, QUILLFELDT P.Evolutionary factors affecting the cross-species utility of newly developed microsatellite markers in seabirds. Molecular Ecology Resources, 2015, 15(5): 1046-1058.
[6]周青利, 王蕊, 张春宵, 周海涛, 易红梅, 王凤华, 李晓辉, 田红丽,葛建镕, 席章营, 王凤格. 玉米 SSR-DNA指纹库构建方案在高粱中的通用性. 玉米科学, 2017: http://kns.cnki.net/kcms/detail/46.1068.S.20170406.1210.020.html.ZHOU Q L, WANG R, ZHANG C X, ZHOU H T, YI H M, WANG F H, LI X H, TIAN H L, GE J R, XI Z Y, WANG F G. A study on universal application of maize SSR-DNA fingerprint database in Sorghum. Journal of Maize Science, 2017: http://kns.cnki.net/kcms/detail/46.1068.S.20170406.1210.020.html. (in Chinese)
[7]魏兴华, 韩斌, 徐群, 黄学辉, 张新明, 龚浩, 冯跃, 堵苑苑, 余汉勇. NY/T 2745-2015, 水稻品种鉴定 SNP标记法. 中华人民共和国农业部, 2015.WEI X H, HAN B, XU Q, HUANG X H, ZHANG X M, GONG H,FENG Y, DU Y Y, YU, H Y. NY/T 2745-2015, Protocol for identification of rice varieties--SNP marker method. The Ministry of Agriculture of the People's Republic of China, 2015. (in Chinese)
[8]徐群, 魏兴华, 庄杰云, 吕波, 袁筱平, 刘平, 张新明, 余汉勇, 堵苑苑. NY/T 1433-2014, 水稻品种鉴定 SSR标记法. 中华人民共和国农业部, 2014.XU Q, WEI X H, ZHUANG J Y, LU B, YUAN Y P, LIU P, ZHANG X M, YU, H Y, DU, Y Y. NY/T 1433-2014, Protocol for identification of rice varieties--SSR marker method. The Ministry of Agriculture of the People's Republic of China, 2014. (in Chinese)
[9]LI L, FANG Z, ZHOU J, CHEN H, HU Z, GAO L, CHEN L, REN S,MA H, LU L, ZHANG W, PENG H. An accurate and efficient method for large-scale SSR genotyping and applications. Nucleic Acids Research, 2017, 45(10): e88.
[10]马艳明, 许勇, 张海英, 张勋基, 陈果, 郭绍贵, 宫国义, 刘志勇,足木热木, 肖菁, 颜国荣. NY/T 2472-2013, 西瓜品种鉴定技术规程 SSR分子标记法, 中华人民共和国农业部, 2013.MA Y M, XU Y, ZHANG H Y, ZHANG X J, CHEN G, GUO S G,GONG G Y, LIU Z Y, ZHU M R M, XIAO J, YAN G Y. NY/T 2472-2013, Identification of watermelon varieties--SSR marker method. The Ministry of Agriculture of the People's Republic of China,2013. (in Chinese)
[11]UNTERSEER S, BAUER E, HABERER G, SEIDEL M, KNAAK C,OUZUNOVA M, MEITINGER T, STROM T M, FRIES R, PAUSCH H. A powerful tool for genome analysis in maize: Development and evaluation of the high density 600K SNP genotyping array. BMC Genomics, 2014, 15: 823.
[12]XU C, REN Y, JIAN Y, GUO Z, ZHANG Y, XIE C, FU J, WANG H,WANG G, XU Y. Development of a maize 55K SNP array with improved genome coverage for molecular breeding. Molecular Breeding, 2017, 37(3): 20.
[13]ELLEGREN H. Microsatellites: simple sequences with complex evolution. Nature Review Genetics, 2004, 5(6): 435-445.
[14]WEBSTER M T, HAGBERG J. Is there evidence for convergent evolution around human microsatellites? Molecular Biology and Evolution, 2007, 24(5): 1097-1100.
[15]BRANDSTR M M, BAGSHAW A T, GEMMELL N J, ELLEGREN H. The relationship between microsatellite polymorphism and recombination hot spots in the human genome. Molecular Biology and Evolution, 2008, 25(12): 2579-2587.
[16]KELKAR Y D, TYEKUCHEVA S, CHIAROMONTE F, MAKOVA K D. The genome-wide determinants of human and chimpanzee microsatellite evolution. Genome Research, 2008, 18(1): 30-38.
[17]FUNGTAMMASAN A, ANANDA G, HILE S E, SU M S, SUN C,HARRIS R, MEDVEDEV P, ECKERT K, MAKOVA K D. Accurate typing of short tandem repeats from genome-wide sequencing data and its applications. Genome Research, 2015, 25(5): 736-749.
[18]ABDULOVIC A L, HILE S E, KUNKEL T A, ECKERT K A. The in vitro fidelity of yeast DNA polymeraseδ and polymeraseɛ holoenzymes during dinucleotide microsatellite DNA synthesis. DNA Repair, 2011, 10(5): 497-505.
[19]BAPTISTE B A, ECKERT K A. DNA polymerase kappa microsatellite synthesis: Two distinct mechanisms of slippagemediated errors. Environmental and Molecular Mutagenesis, 2012,53(9): 787-796.
[20]YANG S, WANG L, HUANG J, ZHANG X, YUAN Y, CHEN J-Q,HURST L D, TIAN D. Parent-progeny sequencing indicates higher mutation rates in heterozygotes. Nature, 2015, 523(7561): 463-467.
