PPARs信号通路在小鼠糖尿病肝病中的作用*
2018-04-02任凯强潘文靖蒋青松
任凯强,薛 莱,黄 波,潘文靖,吴 堃,蒋青松△
(1重庆医科大学药理教研室,重庆市生物化学与分子药理学重点实验室,重庆 400016; 2江油市人民医院临床药学科,四川 江油 621700; 3遵义医学院基础药理教育部重点实验室暨特色民族药教育部国际合作联合实验室,贵州 遵义 563003; 4重庆市人民医院肝胆外科,重庆 400013)
2型糖尿病(type 2 diabetes mellitus,T2DM)是最常见的糖尿病类型,其主要病理特征为胰岛素抵抗,常伴血糖升高和血脂紊乱。肝脏是调节糖、脂代谢最主要的器官,血液中糖脂水平失衡势必影响肝脏的功能,甚至导致肝损伤。已有研究认为,胰岛素抵抗与T2DM和肥胖人群中肝病的发生密切相关[1-2]。在糖尿病患者和肥胖人群中,70%~90%的肝病发生与酒精过量摄取无关,而该比例在普通人群仅为20%~30%[3-4]。作为一种独立的危险因素,糖尿病可导致肝损伤的发生和发展,故把糖尿病并发的肝损伤也称为糖尿病肝病(diabetic hepatopathy),其早期主要表现为非酒精性脂肪性肝病(non-alcoholic fatty liver disease,NAFLD),以脂肪在肝细胞内聚集为主要特征。随着病情进展,NAFLD可能出现肝细胞损伤伴炎症因子产生,发展为非酒精性脂肪性肝炎(non-alcoholic steatohepatitis,NASH),进一步恶化可进展为肝纤维化,甚至出现肝硬化、肝细胞癌或肝功能衰竭等终末期肝病[5]。对于糖尿病引起的肝损伤,目前已经进行了较多研究,但其病理生理基础仍然有待阐明。
过氧化物酶体增殖物激活受体(peroxisome proliferator-activated receptors,PPARs)属于核受体家族成员,其作用与糖脂代谢、脂肪形成、炎症及胰岛素敏感性等生物学功能紧密相关[6]。PPARs有3种亚型,分别为PPARα、PPARβ和PPARγ。目前在糖尿病肝病中的研究主要集中于PPARα在NAFLD的作用,而对于PPARγ的研究较少,PPARβ的作用几乎未见报道,且对于PPARs相关机制在糖尿病肝损伤中的作用缺乏系统研究。另外,目前有研究认为,炎症在糖尿病及其并发症的发生发展中有重要作用。当炎症细胞因子,如肿瘤坏死因子α(tumor necrosis factor α,TNF-α)、白细胞介素(interleukin,IL)-1和IL-6等大量释放,可促使糖尿病肝损伤恶化,从NAFLD进展到NASH[7]。整体动物模型及离体细胞培养均已证实,PPARα、PPARβ和PPARγ可抑制核因子κB(nuclear factor-kappa B,NF-κB)相关信号通路,从而抑制细胞因子如TNF-α、IL-1和IL-6的产生,发挥抗炎作用[8]。作为NF-κB重要的下游因子,环氧合酶2(cyclooxygenase 2,COX-2)及诱导型一氧化氮合成酶(inducible nitric oxide synthase,iNOS)在糖尿病肝病中的作用研究较少。故本实验拟利用高能量饲料喂养联合多次腹腔注射小剂量链脲佐菌素(streptozocin,STZ)诱导小鼠糖尿病模型,对PPARs-炎症相关信号通路在糖尿病肝损伤中的作用进行初步探索。
材 料 和 方 法
1 动物
SPF级4~6周龄雄性昆明小鼠,体重15~20 g,由重庆医科大学实验动物中心提供,许可证号为SCXK(渝)2010-0001。
2 主要试剂
强生稳豪型血糖仪及试纸购自强生(中国)医疗器材有限公司。STZ购自Sigma;胰岛素放免检测试剂盒购自北京北方生物技术研究所;总胆固醇(total cholesterol,TC)、甘油三酯(triglyceride,TG)、丙氨酸转氨酶(alanine aminotransferase,ALT)、天冬氨酸转氨酶(aspartate aminotransferase,AST)和碱性磷酸酶(alkaline phosphatase,ALP)检测试盒均购自南京建成生物技术公司;Trizol购自大连宝生物公司;逆转录试剂盒和SYBR Green Supermix购自Bio-Rad;PCR引物由Invitrogen公司负责合成和纯化;兔抗鼠PPARα和PPARγ多克隆抗体购自Santa Cruz Biotechnology;兔抗鼠PPARβ和COX-2多克隆抗体购自Cayman Chemical Company;兔抗鼠iNOS和β-actin多克隆抗体购自Abcam;兔抗鼠p-NF-κB p65多克隆抗体购自Cell Signaling Technology;山羊抗兔 IgG/HRP Ⅱ抗购自北京中杉金桥生物技术有限公司;其余试剂均为国产分析纯。
3 主要方法
3.1糖尿病肝损伤模型建立小鼠用高糖高脂饲料(成分:10%蔗糖、10%蛋黄、10%猪油、1.5%胆固醇、0.5%胆盐和68%基础饲料混合)喂养4 周后,连续5 d腹腔注射STZ(溶于枸橼酸缓冲液,pH 4.2~4.4,临用前配制,剂量为40 mg·kg-1·d-1)。7 d后,检测小鼠空腹血糖(fasting blood glucose,FBG),FBG≥11.1 mmol/L视为糖尿病形成。糖尿病小鼠继续给予高糖高脂饲料喂养4周后,检测肝脏功能及形态变化,确定糖尿病肝损伤模型建立,即为模型(model)组。正常对照(control)组小鼠给予常规饲料喂养4周后,连续5 d以等量枸橼酸缓冲液腹腔注射,7 d后检测小鼠FBG,然后继续给予常规饲料喂养4周。
3.2生化指标测定眼眶采血,分装后室温静置30 min,4 ℃、3 000×g离心15 min,取血清冻存于-20 ℃冰箱备用。按照各试剂盒说明书操作,ELx800酶标仪(BioTek)分别检测不同波长(TC和TG:490 nm;AST和ALT:510 nm;ALP:520 nm)的吸光度(A)。另外,使用SN-684型放射免疫计数器(上海核所日环光电仪器有限公司)以放免法检测血清中胰岛素含量(insulin,INS),以mU/L表示,并计算胰岛素抵抗指数(HOMA-IR)=FBG×INS/22.5。每组结果来源于8只小鼠。
3.3组织病理学观测实验结束时处死小鼠,分离肝脏,4%的多聚甲醛溶液固定6 h,用不同浓度的乙醇和二甲苯逐级脱水,石蜡包埋,作3~5 μm冠状切片,HE染色,显微镜下观察肝脏组织形态学改变。
3.4qPCR法检测mRNA的表达参照GenBank中小鼠的基因序列合成目的基因引物(表1),用Trizol法提取肝脏组织总RNA,按说明书操作进行逆转录合成cDNA,利用SYBR Green 荧光技术,在Bio-Rad CFX96荧光定量PCR仪按照如下反应条件进行扩增:95 ℃ 30 s; 95 ℃ 5 s,60 ℃ 30 s,40个循环。以β-actin为内参照,Ct值(荧光强度到达阈值时的循环数)为统计参数,相对定量法分析结果。每组重复3次。
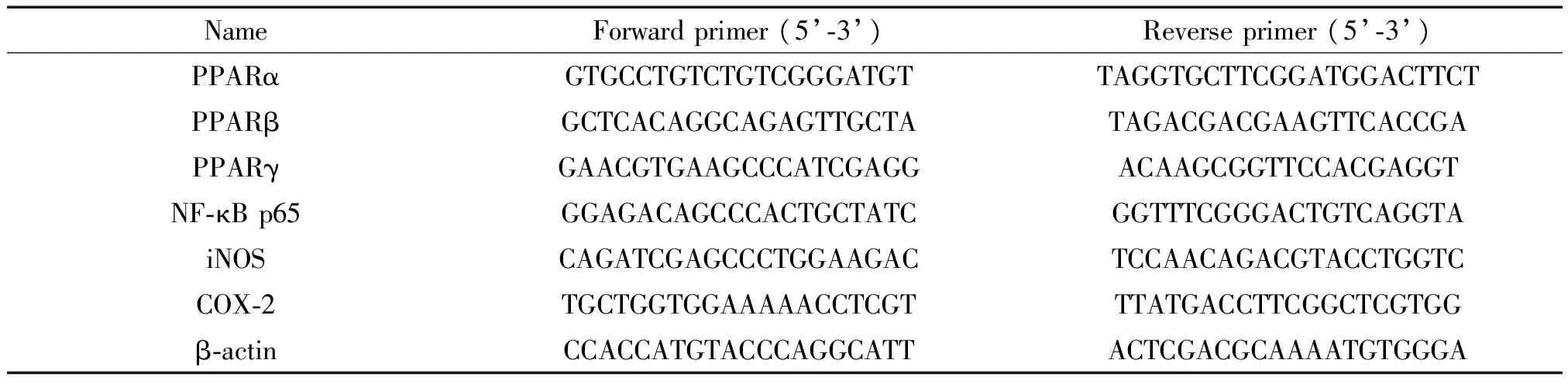
表1 引物序列Table 1.The primer sequences for qPCR
3.5Western blot法检测蛋白的表达取30 μg总蛋白加入含β-巯基乙醇的上样缓冲液,高温煮沸10 min使蛋白变性,行SDS-PAGE,PVDF转膜,BSA封闭1 h,加入不同 I 抗(均1∶1 000稀释),4 ℃过夜,加辣根过氧化物酶标记的 II 抗(1∶5 000稀释)。洗膜后用发光液(Advanstor)显色,凝胶成像仪与Image Lab软件(Bio-Rad)照相显影,分析结果。每组重复3次。
4 统计学处理
用SPSS 20.0统计软件进行统计学分析。实验数据以均数±标准差(mean±SD)表示,组间比较采用t检验,以P<0.05为差异有统计学意义。
结 果
1 小鼠糖尿病相关指标的变化
给予STZ 7 d后,与对照组相比,模型组小鼠FBG显著升高,从(5.20±0.25) mmol/L升高至(20.53±1.30) mmol/L (P<0.01),远大于11.1 mmol/L,说明糖尿病形成;糖尿病形成4周后,模型组小鼠FBG升至(23.63±1.77) mmol/L,与同期正常对照组的FBG[(4.74±0.59) mmol/L]比较,升高了4.0倍(P<0.01)。糖尿病肝损伤模型组小鼠的血清胰岛素水平亦明显升高,为正常对照组的4.2倍(P<0.01)。同时,胰岛素抵抗指数HOMA-IR增加了18.0倍(P<0.01),见图1。

Figure 1.The levels of insulin and HOMA-IR in diabetic hepatopathy mice.Mean±SD.n=8.**P<0.01vscontrol group.
图1糖尿病肝损伤小鼠血清胰岛素和胰岛素抵抗指数水平的变化
2 小鼠血脂及肝脏功能和形态的变化
与正常组相比,糖尿病肝损伤模型组小鼠的TC和TG均显著升高(P<0.01)。同时,肝功能指标ALT、AST和ALP分别亦显著增加(P<0.01),见表2。组织病理学检测显示,糖尿病形成4周后,糖尿病小鼠肝组织着色浅,肝小叶结构消失,肝细胞排列明显紊乱,肿胀明显,胞浆内可见大小不一脂滴,局部伴炎症细胞浸润,细胞内出现大量脂肪空泡,见图2。

表2 小鼠血脂和肝功能的变化Table 2.The levels of TC,TG,ALT,AST and ALP in diabetic hepatopathy mice (Mean±SD.n=8)
**P<0.01vscontrol group.

Figure 2.The hepatic histopathology in diabetic hepatopathy mice (HE staining,×200).
图2糖尿病肝损伤小鼠肝组织病理变化
3 糖尿病肝损伤小鼠肝脏PPARs mRNA和蛋白表达的变化
在糖尿病肝损伤小鼠中,其肝脏PPARα、PPARβ和PPARγ的mRNA和蛋白表达均明显下调(P<0.05或P<0.01),见图3。

Figure 3.The expression of PPARα,PPARβ and PPARγ at mRNA and protein levels in the livers of diabetic he-patopathy mice.Mean±SD.n=3.*P<0.05,**P<0.01vscontrol group.
图3糖尿病肝损伤小鼠肝脏PPARα、PPARβ和PPARγmRNA和蛋白表达的变化
4 糖尿病肝损伤小鼠肝脏NF-κB p65、iNOS和COX-2 mRNA和蛋白表达的变化
在糖尿病肝损伤小鼠,其肝脏的NF-κB p65、iNOS和 COX-2的mRNA和蛋白表达均明显上调(P<0.01),见图4。

Figure 4.The expressions of NF-κB p65,COX-2 and iNOS at mRNA and protein levels in the livers of diabetic he-patopathy mice.Mean±SD.n=3.**P<0.01vscontrol group.
图4糖尿病肝损伤小鼠肝脏NF-κBp65、COX-2和iNOSmRNA和蛋白表达的变化
讨 论
肝损伤是糖尿病严重并发症之一,具有较高的发病率和死亡率,严重影响了人类的生命健康和生活质量。糖尿病肝损伤临床症状表现较少且不典型,目前其发病机制尚未阐明。众所周知,胰岛素除了促进糖原的合成和贮存,加速葡萄糖的氧化酵解,抑制糖原分解和糖异生外,还可促进脂肪合成,抑制脂肪分解,减少游离脂肪酸和酮体生成,增加脂肪酸和葡萄糖的转运。糖尿病时由于胰岛素抵抗,肝脏对胰岛素作用的敏感性下降,导致肝脏葡萄糖排出增加和机体对葡萄糖的摄取及利用减少,TG合成增加;同时,外周脂肪组织裂解增加,肝脏脂肪酸的从头合成途径显著提高,但其脂肪酸氧化及输出较低,从而引起脂肪酸在肝脏堆积[9]。另外,胰岛素抵抗时极低密度脂蛋白(very-low-density lipoprotein,VLDL)分泌减少,也进一步增加肝脏脂肪聚集,引起NAFLD发生[1]。而大量脂质堆积,诱导肝脏活性氧族(reactive oxygen species,ROS)、IL-6、C反应蛋白等促炎因子增多,引起持续炎症反应,使NAFLD恶化进展为NASH[10]。
本研究中,在高糖高脂饲料喂养联合连续多次小剂量给予STZ后,模型组小鼠FBG水平一直高于11.1 mmol/L,提示糖尿病形成。糖尿病小鼠继续予高能量饲料喂养4周,其血清胰岛素水平和胰岛素抵抗指数明显增加;TC和TG水平升高,提示小鼠出现了严重的胰岛素抵抗并伴随体内血糖、血脂水平异常。同时,模型组小鼠ALT、AST和ALP均显著升高,提示其肝功能严重受损。组织形态学检查显示,在糖尿病形成4周后,出现肝细胞脂肪变性、炎性细胞浸润等糖尿病肝病的典型表现。上述生化指标结合组织病理学改变,提示糖尿病小鼠出现肝损伤的并发症。
PPARs家族是调控代谢平衡、糖、脂和能量代谢及胰岛素敏感性等的重要转录因子[6]。其中,PPARα主要通过调控脂肪酸代谢相关基因来调节脂类代谢,包括脂肪酸的β氧化及运输过程等,从而使TG、游离脂肪酸和 VLDL合成减少。PPARβ调节基因表达参与细胞的能量代谢、脂质和葡萄糖的利用,维持机体能量平衡。PPARγ调控参与脂肪前体细胞分化的多个基因的转录,调节胰岛素介导的外周组织对葡萄糖的摄取,增加胰岛素敏感性[11]。另外,炎症细胞因子也是PPARs作用的靶标之一。整体动物模型及离体细胞培养均已证实,PPARα、PPARβ和PPARγ具有明显的抗炎作用[12]。PPARs激活后,通过作用于炎症相关转录因子,如NF-κB,最终使趋化因子、细胞因子及黏附分子等的生成减少,产生抗炎作用,从而减轻组织损伤[13-14]。本实验结果发现,在糖尿病肝损伤小鼠,其肝脏PPARα、PPARβ及PPARγ mRNA及蛋白表达均明显下调,而NF-κB的表达明显升高,提示PPARs-NF-κB相关信号通路异常在糖尿病肝病中可能有重要作用。
NF-κB作为一种重要的核转录因子,是近年研究的热点。NF-κB调控着多种基因的表达,在免疫反应、细胞凋亡和肿瘤的发生发展,尤其是炎症反应中扮演着重要角色。NF-κB激活后导致大量促炎症细胞因子,如IL-1、IL-2、IL-6、IL-12和TNF-α等生成,使炎症更进一步加重。在NF-κB众多的下游因子中,COX-2和iNOS属于诱导型效应酶,在大多数组织中基本不表达,但在一些炎症因子、生长因子及促癌剂等的作用下表达增强。COX-2和iNOS基因启动子区域含NF-κB结合位点,NF-κB可通过作用于这些结合位点调控COX-2和iNOS表达,激活NF-κB可激活COX-2和iNOS基因启动子活性,上调两者的基因转录和蛋白表达,参与多种病理生理过程[15]。我们前期研究表明[16],NF-κB-COX-2/iNOS的激活参与了糖尿病心肌肥厚的损伤过程。那么,该通路在糖尿病肝损伤过程中是否也有类似的作用?在不同方法建立的NAFLD模型,其肝脏NF-κB、COX-2和iNOS表达都明显上调[17-18]。本研究结果与之一致,在糖尿病肝病小鼠肝脏,随着NF-κB mRNA和蛋白表达的增加,COX-2和iNOS的表达也随之增加。这些结果提示,NF-κB-COX-2/iNOS可能也参与了糖尿病肝病的炎症损伤过程。
本研究结果提示糖尿病肝病的发生可能与PPARs-NF-κB-COX-2/iNOS信号通路异常有关。糖尿病时胰岛素抵抗及血糖、血脂水平紊乱,PPARα、PPARβ及PPARγ表达下调,NF-κB活性增加,其下游炎症因子COX-2和iNOS也随之被激活,使NAFLD向NASH进展,加重肝损伤。因此,PPARs-NF-κB-COX-2/iNOS信号通路可能是糖尿病肝损伤防治的重要干预靶点之一,激活PPARs或抑制NF-κB-COX-2/iNOS可能都有防治糖尿病肝病的作用。但糖尿病肝损伤的影响因素较为复杂,对于PPARs-炎症相关信号通路在其中的作用还需进行更多深入研究。
[参考文献]
[1]Bhatt HB,Smith RJ.Fatty liver disease in diabetes mellitus[J].Hepatobiliary Surg Nutr,2015,4(2):101-108.
[2]Polimeni L,Del Ben M,Baratta F,et al.Oxidative stress:new insights on the association of non-alcoholic fatty liver disease and atherosclerosis[J].World J Hepatol,2015,7(10):1325-1336.
[3]Vernon G,Baranova A,Younossi ZM.Systematic review:the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults[J].Aliment Pharmacol Ther,2011,34(3):274-285.
[4]Kemmer N,Neff GW,Franco E,et al.Nonalcoholic fatty liver disease epidemic and its implications for liver transplantation [J].Transplantation,2013,96(10):860-862.
[5]Sumida Y,Seko Y,Yoneda M.Novel antidiabetic medications for non-alcoholic fatty liver disease with type 2 diabetes mellitus[J].Hepatol Res,2017,47(4):266-280.
[6]Gross B,Pawlak M,Lefebvre P,et al.PPARs in obesity-induced T2DM,dyslipidaemia and NAFLD [J].Nat Rev Endocrinol,2017,13(1):36-49.
[7]Perumpail RB,Liu A,Wong RJ,et al.Pathogenesis of hepatocarcinogenesis in non-cirrhotic nonalcoho-lic fatty liver disease:Potential mechanistic pathways[J].World J Hepatol,2015,7(22):2384-2388.
[8]Monsalve FA,Pyarasani RD,Delgado-Lopez F,et al.Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases[J].Mediators Inflamm,2013,2013:549627.
[9]瞿玉兰,韩泽广.脂代谢紊乱在非酒精性脂肪肝发病中的作用[J].生命科学,2017,47(5):496-507.
[10] Kitade H,Chen G,Ni Y,et al.Nonalcoholic fatty liver disease and insulin resistance:new insights and potential new treatments[J].Nutrients,2017,9(4):E387.
[11] 张秀红,宣姣,亓志刚.PPARα、γ和δ:胰岛素抵抗治疗的靶点[J].中国生物化学与分子生物学报,2014,30(6):543-548.
[12] Derosa G,Sahebkar A,Maffioli P.The role of various peroxisome proliferator-activated receptors and their li-gands in clinical practice[J].J Cell Physiol,2018,233(1):153-161.
[13] Palomer X,Salvadó L,Barroso E,et al.An overview of the crosstalk between inflammatory processes and metabo-lic dysregulation during diabetic cardiomyopathy[J].Int J Cardiol,2013,168(4):3160-3172.
[14] 李秀丽,王晓晖.PPAR激动剂在治疗非酒精性脂肪肝病重的应用前景[J].中国医院药学杂志,2015,35(23):2153-2156.
[15] Tak PP,Firestein GS.NF-κB:a key role in inflammatory diseases[J].Clin Invest,2001,107(1):7-11.
[16] 黄波,薛莱,吴阳,等.虎杖苷对小鼠糖尿病心肌肥厚的保护作用[J].中国中药杂志,2015,40(21):4256-4261.
[17] Xiao J,Ho CT,Liong EC,et al.Epigallocatechin gallate attenuates fibrosis,oxidative stress,and inflammation in non-alcoholic fatty liver disease rat model through TGF/SMAD,PI3K/Akt/FoxO1,and NF-κB pathways[J].Eur J Nutr,2014,53(1):187-199.
[18] Tipoe GL,Ho CT,Liong EC,et al.Voluntary oral fee-ding of rats not requiring a very high fat diet is a clinically relevant animal model of non-alcoholic fatty liver disease (NAFLD)[J].Histol Histopathol,2009,24(9):1161-1169.
