Absolute configuration of curdione and its three isomers by NMR, ECD and DFT calculations: an insight into the scope of unsaturated ketone helicity rule based on an ECD study
2018-04-02FeitingZhangYuxinLiTianhuiZhuWenheZhangBinQinSongYou
Feiting Zhang, Yuxin Li, Tianhui Zhu, Wenhe Zhang, Bin Qin*, Song You*
School of Life Sciences & Biopharmaceutical Sciences, Shenyang Pharmaceutical University,Shenyang 110016, China
1 Introduction
Curdione [(4S,7S)-curdione, Cd1, Fig. 1],a sesquiterpenoid natural product with a flexible ten member ring and two stereogenic centers, was isolated from Curcuma wenyujin and related species which had long been used as a remedies for cervical cancer in mainland China [1,2]. It transannular cyclization product, curcumalactone [3],upon acid treatment, possessing a spirolactone skeleton had been total synthesised by Romo and co-workers in 2012 [4]. Our group obtained a series of novel derivatives of curdione, curcumalactone and their isomers, 16 pairs of the mirror-image compounds [5,6]. However, the absolute configuration was laboriously determined by the circumstantial evidences of MS,1H,13C NMR, ECD data and the literatures [3,5-7]. In this paper, the density functional theory calculations were used to offer a convenient, reliable, fast method for the assignment of absolute configuration.
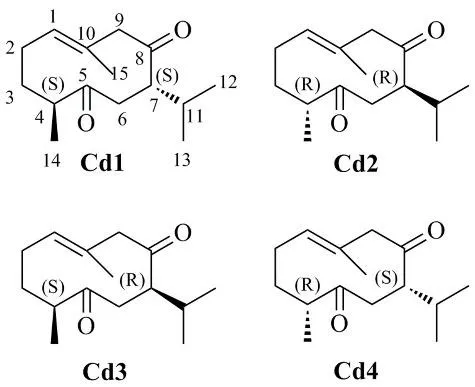
Fig. 1 Curdione (Cd1) and its stereoisomers
Cd1 and (4S,7R)-curdione (Cd3) are the sesquiterpenoid nature products. (4R,7R)-curdione (Cd2) and (4R,7S)-curdione (Cd4) are enantiomers of naturally occurring Cd1 and Cd3,respectively, which were semi-synthesized from their above precursors. All these four compounds have the β,γ-unsaturated ketone group. However,the relationship between the experimental ECD spectra and the stereostructures of these compounds almost exclusively coincided with the empirical α,βunsaturated ketone helicity rule [8-10], which the positive (negative) O=C-C=C torsion angle is shown to respond to the positive (negative) sign of the CE(Fig. 2). This rule has been repeatedly used as a tool in the determination of the absolute configuration of a vast range of compounds containing the α,βunsaturated ketone chromophore [11]. Unfortunately,the application to the compounds containing β,γunsaturated ketone group is not reported. In an attempt to explain the reason for the compounds with the β,γ-unsaturated ketone group can be applied to the α,β-unsaturated ketones helicity rule, TDDFT calculations were carried out to elucidate the relationship between torsion angle and the ECD band.
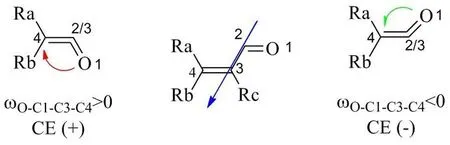
Fig. 2 Graphical representation of the α,β-unsaturated ketone helicity rule
2 Material and methods
Cd1 and Cd3 were isolated from the essential oil of C. wenyujin by column chromatography with petroleum ether−EtOAc (50:1, v/v). Their enantiomers Cd2 and Cd4 need to be semisynthesized. And the synthesis of them were performed following the reported approach by isomerization at C-4 of natural Cd3 and Cd1 via a three-step reaction [5], respectively. The identification of these four compounds were carried out by the experimental and theoretical NMR and ECD spectra. The NMR spectra of all molecules were recorded on a Bruker AV-600 NMR spectrometer at usual probe temperature, employing tetramethylsilane (TMS) as internal reference. The operating frequencies were 600 MHz and 150 MHz for1H and13C, respectively. All measurements were done using deuterated trichloromethane as solvent.The experimental ECD spectra were measured in MeOH on a JASCO CD-2095 circular dichroism chiral detector at the 220-400 nm range.
Curdione and its isomers conformational analysis were performed by the Spartan 10 package [12] using the MMFF94s molecular mechanics force field with a Systematic and Monte Carlo search. The conformers with a Boltzmann population above 1% were further fully subjected to further optimize in the Gaussian 09 package [13] at DFT/B3LYP/TZVP level. All conformers were real minima, as no imaginary vibrational frequencies were found. Free energies were calculated and used to determine the Boltzmann populations of the conformers at 298.15 K.
NMR chemical shift calculations were obtained with the GIAO method at the B3LYP/6-311++G(d,p) level on the stable conformers. TMS, calculated at the same level of theory, was used as reference to scale the absolute shielding value. The theoretical NMR spectra were obtained as weighted averages of Boltzmann populations.
Calculations of ECD spectra were performed at the TDDFT/CAM-B3LYP/CPCM(MeOH)/augcc-pVDZ level. The theoretical ECD were obtained as weighted averages of Boltzmann populations.The ECD spectra were obtained from calculated excitation energies and rotational strengths as a sum of Gaussian functions centered at the wavelength of each transition with a parameter σ (width of the band at half-height) of 0.2 eV and elaborated using the SpecDis v1.53 program [14]. Specifically, the TDDFT/CAM-B3LYP/aug-cc-pVTZ level was employed for compound Cd1. Meanwhile, Cd1 was employed the IEFPCM as the solvent model.
3 Results
3.1 Geometrical structures
The structures of curdione and its isomers here studied were showed in Fig. 1. They possess two stereogenic centers and differ in the position C-4 of the methyl substituent and the position C-7 of isopropyl substituent in the ten member ring. From the early research [15,16], it was clearly reasonable that the dihedral angles of φC4-C5-C6-C7and φC9-C10-C1-C2were 180° (±20°) and the substituents of C-10 and C-5 were approximately perpendicular to the average plane of the molecular. Owing to the energy need for conformational inversion of the ten member ring was higher, the four principal conformations were designated as anti-up, syn-up, anti-down,syn-down (Fig. S1) for all molecules at the mercy of our consideration. Based on the four principal conformations, a detail conformation searching for each principal conformation of each molecule were carried out at the molecular mechanics (MM)level, and described above yielded 89, 260, 99 and 78 conformers for the Cd1, Cd2, Cd3 and Cd4,respectively. Finally, from the resulting unique minima, only the geometries with a Boltzmann population above 1% were retained for further optimized using the B3LYP functional and the TZVP basis set in vacuo. The relative energies and the populations of all conformers were given in Table S1. Cd1 and Cd2 both led to 3 unique conformers with a Boltzmann population above 1% (Figs. S2 and S3). Cd1a and Cd2a are the dominant conformers of Cd1 and Cd2, respectively, which occupy 96.01%and 95.44% population. Cd3 and Cd4 each have five conformers with a Boltzmann population above 1%,where three conformers occupy greater than 10%population of abundance of either compound (Figs.S4 and S5).
3.2 NMR studies
The1H and13C NMR chemical shifts indicated that Cd1 had the same δHand δCvalues with Cd2,and so did Cd3 with Cd4, which may account as enantiomers of each other (Fig. S6, Fig. S7). In1H NMR spectrum, there are 24 hydrogen atoms attached in each molecule of Cd1-Cd4 (Fig. S6).Generally, the chemical shift of methyl, methylene and methyne can be observed at approximately 0.87,1.20 and 1.55, respectively. But, these chemical shift values may change, due to proton's electronic environment. That is, if electron accepting atoms or groups directly attached or closed, they could reduce the shielding. And so, the hydrogen atom attached to an electron-withdrawing atom or group could move towards to a higher frequency. In compound Cd1 or Cd2, the proton chemical shifts of isopropyl group were observed at 1.85, 0.94 and 0.87, and the experimental values of the two methyl groups were recorded as 1.64 and 0.97, respectively.The chemical shift of other hydrogens was found higher than the forementioned hydrogens due to the carbonyl groups and the carbon-carbon double bond. Based on the degree of the influences of these groups and the calculated data, we can easily decide which one is which. With the same analysis, we can also decide the hydrogens chemical shifts in Cd3 or Cd4. In13C NMR spectrum, the expected 15 different carbon atoms can be seen at a glance (Fig.S7). From the early research and the calculated data,the peaks of those carbon atoms also decided.
The NMR chemical shift calculations were obtained with the GIAO method at the B3LYP/6-311++G(d,p) level on the stable conformers. TMS,calculated at the same level of theory, was used as reference to scale the absolute shielding value [17].The theoretical1H chemical shifts were carried out from a weighted sum of the stable conformers by Boltzmann population (Table 1), which made good overall qualitative agreement with the experimental data. The average absolute errors (AAEs) of the theoretical1H values obtained in CDCl3were low,0.07 for Cd1 and Cd2, and 0.06 for Cd3 and Cd4.The largest errors in Cd1 and Cd2 were observed at the H-6a, H-6b, H-9a and H-9b, and in Cd3 and Cd4 were observed at the H-4 and H-6a, H-6b.Both of these errors were possibly due to the lack of the influence by solvent molecules near the carboxyl group. The largest differences between the diastereomers were observed for H-3a, H-3b and H-4. The experimental and theoretical Δδ values for these were (experiment; theory) H-3a (0.2; 0.21),H-3b (0.13, 0.3), H-4 (0.18; 0.01).

Table 1 1H NMR assignments for Cd1, Cd2, Cd3 and Cd4

Continued Table 1
Table 2 showed the theoretical13C NMR data of Cd1-Cd4 result from a weighted sum of quite possible conformers by Boltzmann population,which can also find that Cd1 had the same δCvalues with Cd2 and so did Cd3 with Cd4. Compared with the experimental spectra, the theoretical13C shift were reasonably well agreed, with the AAEs of 3.36 for Cd1 and Cd2, 3.29 for Cd3 and Cd4.In compound Cd1 or Cd2, the largest error was observed at the C-5, possibly due to the lack of explicit hydrogen bonding from solvent molecules to the carboxyl group. And in Cd3 or Cd4, the largest errors were observed at the C-3 and C-4, possibly due to the flexibility of the ring. In the experimental13C spectra, C-6, C-8, and C-15 exhibited the most substantial differences between the diastereomers.The ∆δ values for the diastereomers were(experiment; theory) C-6 (2.2; 3.95), C-8 (1.7;0.08), C-15 (1.7; 3.4). However, the theory also predicted some large Δδ for C-5 (3.59) and C-9(3.47), not supported by experiment (0.8) and (0.6).Overall, the Δδ values were small compared to the absolute values of the carbon shifts, which is not surprising as13C nuclei are relatively electron rich and their chemical shifts are therefore dominated by the chemical bonds formed with neighboring nuclei. Notwithstanding some small discrepancies,the diastereomers can be separated easily by the experimental and theoretical1H and13C spectra.
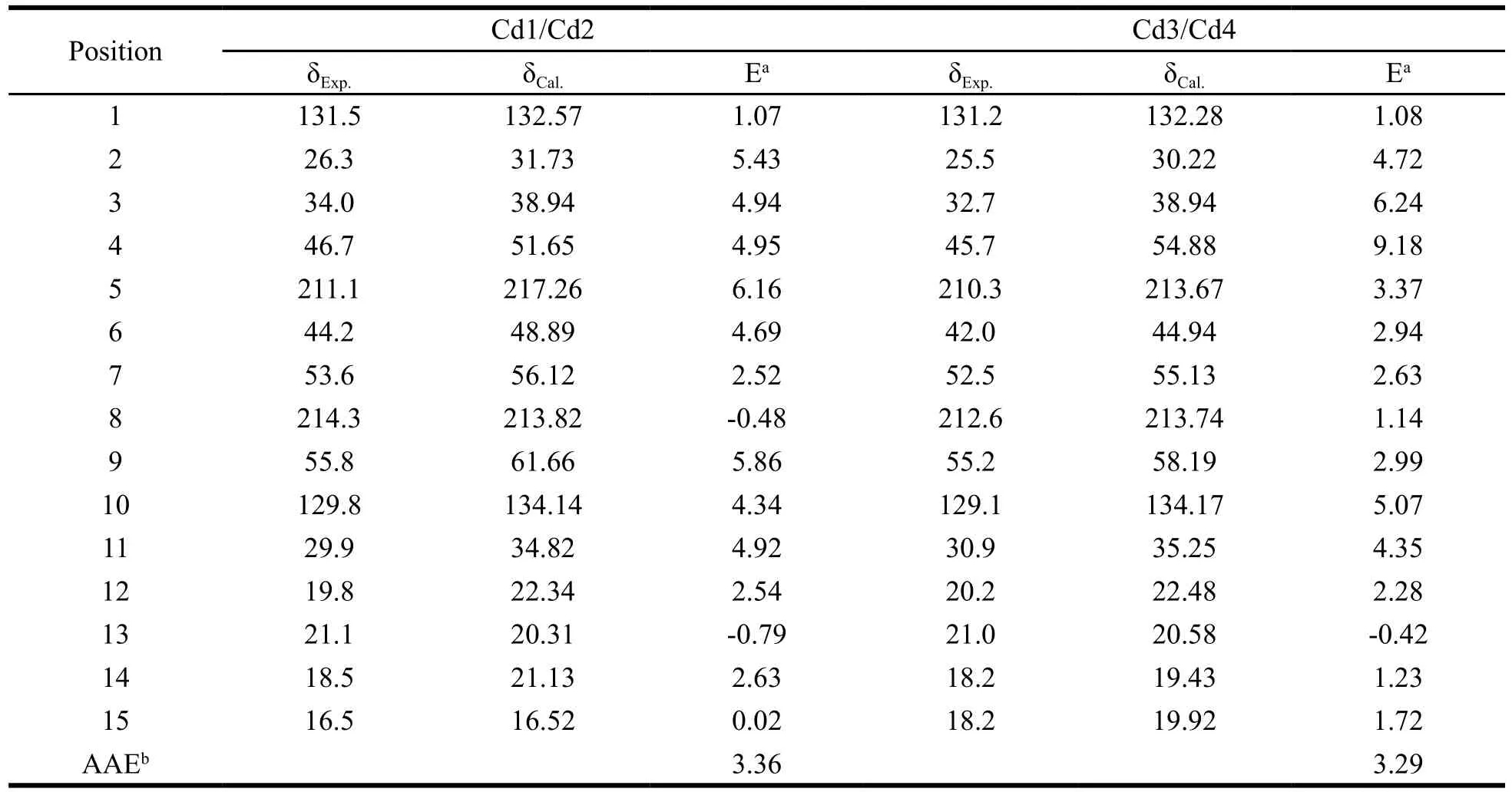
Table 2 13C NMR assignments for Cd1, Cd2, Cd3 and Cd4
3.3 ECD studies
On the enantiomers, the ECD spectra were used to distinguish them from each other. In the experimental ECD spectra of those four compounds,two cotton bands could be observed near 220 nm and 296 nm of all compounds (Fig. 3). ECD spectra of Cd1 and Cd2 were good mirror images of one another (Fig. 3A), indicated that these two compounds were enantiomers. However, the spectra of Cd3 and Cd4 were no more global mirror symmetry like the spectra of Cd1 and Cd2 (Fig.3B). In Cd3 and Cd4, the expected cotton bands at 220 nm were not gone by contraries, both of them had a positive rotation. The reason for the compound Cd4 have a positive rotation at 220 nm may be the solvent effect, that the solute and the MeOH molecule are interacted with a hydroxyl group.
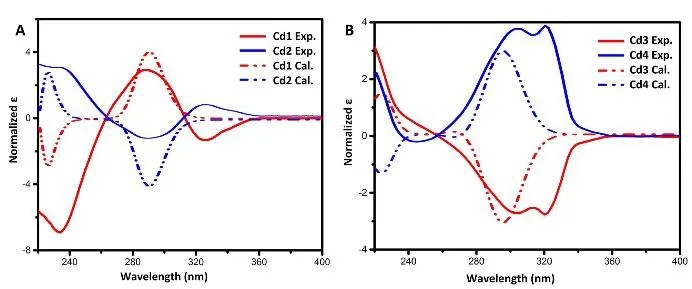
Fig. 3 Comparison of experimental and theoretical ECD spectra for curdione and its isomers
The theoretical ECD spectra were performed at the TDDFT/CAM-B3LYP/CPCM(CH3OH)/augcc-pVDZ level on the most populated conformers.Theoretical ECD spectra of Cd1 and Cd2 depicted in Fig. 3A, Cd3 and Cd4 depicted in Fig. 3B, were good mirror images with each other, which exhibited two oppositely rotations near 296 nm and 220 nm.More specifically, Cd1 gave a positive rotation at 296 nm and a negative rotation at 220 nm, Cd2 gave the opposite rotations as Cd1. Meanwhile, Cd3 gave a negative rotation near 296 nm and a positive rotation at 220 nm, Cd4 gave the opposite rotations as Cd3. Comparison between the calculated and experimental ECD spectra of those four compounds showed a satisfactory match except for the cotton band at 220 nm of Cd4. In the compounds Cd1 and Cd2, the ECD spectra of individual conformers indicated that the transition at 296 nm was contributed by the dominant conformer (Fig. S8). As inferred from MO analysis of Cd1, CEs at 296 nm could be ascribed to n-π* transition of the β,γunsaturated ketone group (Fig. S9). Meanwhile, the calculated ECD spectra of Cd3 and Cd4 had two transitions, at 220 nm and 296 nm. Comparing with the spectra of individual conformers and the average ECD spectra (Fig. S10), we found that the three most populated conformers have great contribution to the average ECD spectra of the CE at 296 nm,which may give the reason why the experimental spectra had a broad peak near 300 nm. With the MO analysis of Cd3, the transition at 296 nm could be also ascribed to n-π* transitions of the β,γunsaturated ketone group (Fig. S11). In combination with NMR, ECD provided the absolute configuration for the stereogenic centers of C-4 and C-7, which had yield full absolute configuration assignment about curdione and its three isomers.
To assess the reliability of theoretical ECD spectra, we performed TDDFT computations with more advanced TDDFT/CAM-B3LYP/aug-cc-pVTZ level for Cd1. Compared with the aug-cc-pVDZ level, this level had the similar features (Fig. S12),which means the aug-cc-pVDZ level is good enough.To account for the effect of the MeOH solvent on conformational stability, we also employed the integral equation formalism polarizable continuum model (IEFPCM) as the solvent model in Cd1.In this case, the calculated ECD spectrum give a satisfactory match with the spectrum of ECD calculated by the CPCM model (Fig. S13).
3.4 Helicity rule studies
During the ECD analysis, we found the relationship between the ECD spectra and the stereostructures of curdione and its isomers almost exclusively coincided with the empirical α,βunsaturated ketone helicity rule, which together with ketone octant rule [18,19] and exciton chirality rule [20] have for years been the most popular and widespread method to assign the absolute configuration of organic molecules without ECD calculations. Therefore, the dominant conformers of the four compounds were envisaged to test applicability of the α,β-unsaturated ketone helicity rule to a case of β,γ-unsaturated ketone group with a n-π* transition. The relationship between the n-π*ECD band and the torsion angle ωO-C8-C10-C1 was depicted in Fig. 4. In the conformers Cd1a and Cd4a,a positive torsion angle is respect to a positive sign of CE. And Cd2a and Cd3a with a negative torsion angle have a negative sign of CE. The relationship was appreciable that positive (negative) torsion angle is respect to the positive (negative) sign of CE by the compounds with β,γ-unsaturated ketone group,which is called the unsaturated ketone helicity rule.

Fig. 4 The relationship between the sign of CEs and the torsion angle ωO-C8-C10-C1 of the dominant conformers of curdione and its isomers
In order to establish a priori and resultant application of the unsaturated ketone helicity rule,more conformers were carried out to elucidate the relationship between torsion angle and the ECD band. The conformers were obtained by Monte Carlo search and further optimized at the B3LYP/TZVP level in vacuo, which labeled as a-p (Fig.S14). Further, the theoretical ECD spectra of these conformers were calculated at TDDFT/CAMB3LYP/CPCM(MeOH)/aug-cc-pVDZ level (Fig.S15). After the calculations, the torsion angles and the sign of CEs were analyzed for all the conformers. Fortunately, the relationship between torsion angle and the ECD band is fully consistent with the unsaturated ketone helicity rule (Table 3).Table 3 also shows that the longer the distance between C-8 and C-10 is, the weaker the rotatory strength of ECD band is. And the weakest rotatory strength observed is conformer p, which the distance value is 2.55Å. Such analysis demonstrates that the application of the unsaturated ketone helicity rule as follow: 1) the unsaturated ketone group should possess an electronic transition which can be unequivocally assigned to a n-π* transition; 2) the distance between the C (C=O) and C (C=C) can be used as the condition to estimate the application of the unsaturated ketones helicity rule, which should be less than 2.55Å; 3) the sign of the n-π* CE is governed by the helicity of the system, i.e. positive(negative) sign of the CE corresponds to the positive(negative) torsion angle.
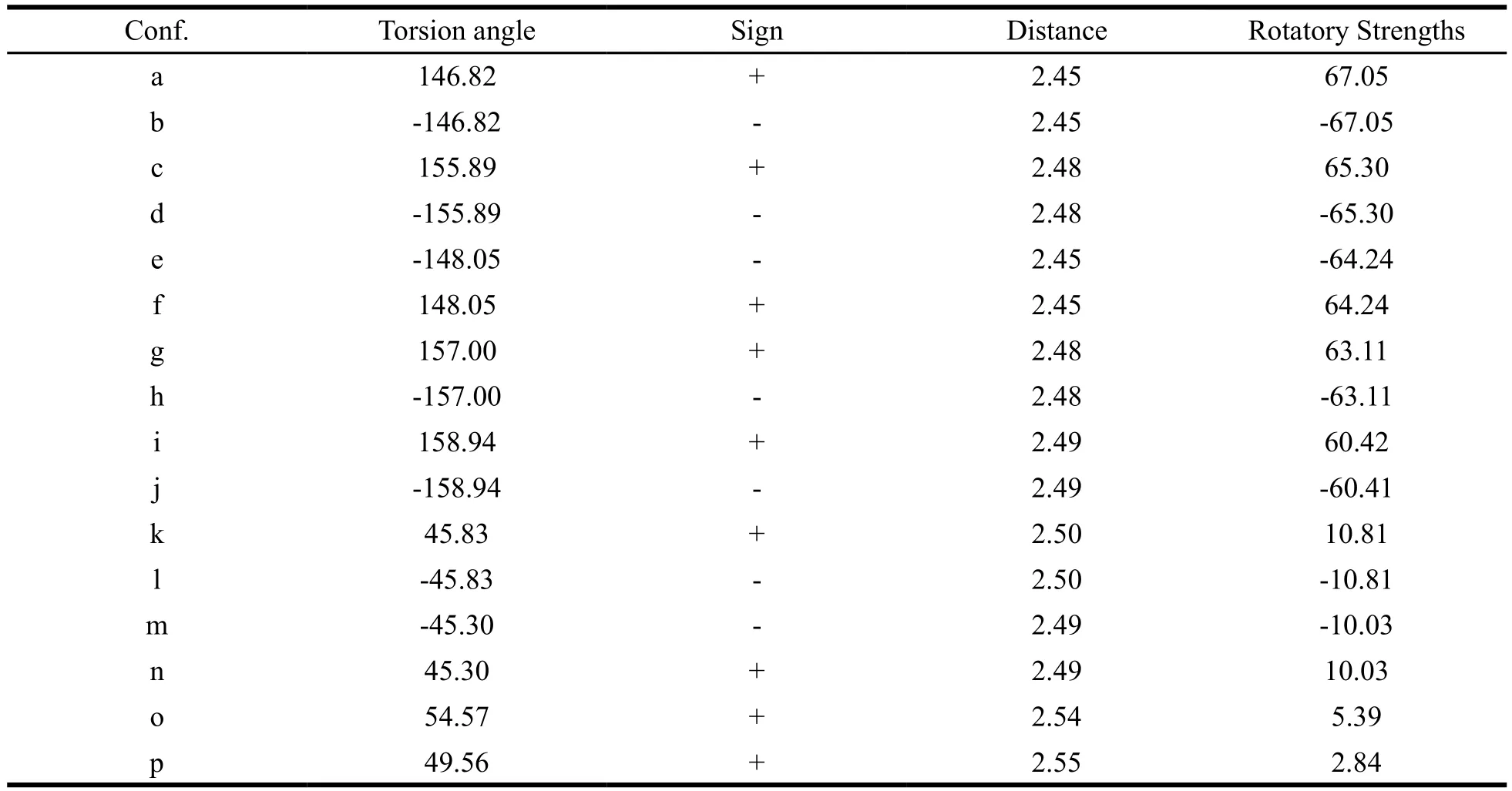
Table 3 The relationship between the sign of CEs and the stereostructures of the conformers a-p
4 Conclusions
In the present studies, the absolute configurations of curdione and its three isomers were confirmed by the experimental and theoretical NMR,ECD spectra. And the curdione skeleton offered the unique opportunity to elucidate the applicability of the α,β-unsaturated ketones helicity rule to the unsaturated ketone groups with the distance between the two unsaturated carbon atoms less than about 2.55Å. It was demonstrated that the NMR and ECD calculations could provide a suitable method to assign the absolute configuration for flexible molecules and the mere application of unsaturated ketones helicity rule would be ambiguous.
Supplementary Material
All the experimental and theoretical ECD spectra and1H-NMR,13C-NMR spectra of the various conformers of the Cd1-Cd4.
[1] Chinese Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China. 1 (2015). China Medical Science Press, 2015, 274-275.
[2] Aggarwal B, Yuan W, Li S, et al. Curcumin-free turmeric exhibits anti-inflammatory and anticancer activities:Identification of novel components of turmeric. Mol Nutr Food Res, 2013, 57: 1529-1542.
[3] Hu JH, Han XW, Ji T, et al. Formation of curcuma lactone and determination of its molecular-structure.Kexue Tongbao, 1987, 32: 816-820.
[4] Leverett CA, Purohit VC, Johnson AG, et al. Dyotropic rearrangements of fused tricyclic β-lactones:application to the synthesis of (-)-curcumanolide A and (-)-curcumalactone. J Am Chem Soc, 2012, 134:13348-13356.
[5] Qin B, Li Y, Meng L, et al. “Mirror-image” manipulation of curdione stereoisomer scaffolds by chemical and biological approaches: development of a sesquiterpenoid library. J Nat Prod, 2015, 78: 272-278.
[6] Chen Y, Zhang L, Qin B, et al. An insight into the curdione biotransformation pathway by Aspergillus niger. Nat Prod Res, 2014, 28: 454-460.
[7] Ohkura T, Gao JF, Harimaya K, et al. Determination of Stereostructure of Neocurdione Isolated from Curcuma wenyujin. The Japanese journal of pharmacognosy,1986, 40: 352-354.
[8] Djerassi C, Records R, Bunnenberg E, et al. Inherently dissymetric chromophores. Optical rotatory dispersion of α, β-unsaturated ketones and conformational analysis of cyclohexenones. J Am Chem Soc, 1962, 84: 870-872.
[9] Wu S, Yang N, Yang L, et al. A novel type of optically active helical polymers: Synthesis and characterization of poly (α, β-unsaturated ketone). J Polym Sci Pol Chem, 2010, 48: 1441-1448.
[10] Kuball HG, Neubrech S, Schönhofer A. Optical activity of oriented molecules. α, β-unsaturated steroid ketones and their sector rules. Chem Phys, 1992, 163: 115-132.
[11] Cerda-García-Rojas CM, Coronel AC, de Lampasona MEP, et al. Absolute configuration of lippifoliane and africanane derivatives. J Nat Prod, 2005, 68: 659-665.
[12] Deppmeier B J, Driessen A J, Hehre T S, et al. Spartan 10. Wavefunction Inc.: Irvine, CA, 2011.
[13] Frisch M, Trucks GW, Schlegel HB, et al. Gaussian 09,revision D. 01, Gaussian Inc. Wallingford, CT, USA,2009.
[14] Bruhn T, Hemberger Y, Schaumlöffel A, et al. SpecDis,Version 1.53; University of Wuerzburg: Wuerzburg,Germany, 2011.
[15] Inayama S, Gao JF, Harimaya K, et al. The absolute configuration of curdione and the stereostructure of curcumalactone from Curcuma wenyujin. Chem Pharm Bull, 1985, 33: 2179-2182.
[16] Harimaya K, Ohkura T, Gao JF, et al. Conformational analysis of curdione and neocurdione. Chem Pharm Bull, 1987, 35: 3866-3869.
[17] Karabacak M, Kose E, Kurt M. FT-Raman, FT-IR spectra and DFT calculations on monomeric and dimeric structures of 5-fluoro-and 5-chloro-salicylic acid. J Raman Spectrosc, 2010, 41: 1085-1097.
[18] Lightner DA, Chang TC. Octant rule. III. Experimental proof for front octants. J Am Chem Soc, 1974, 96:3015-3016.
[19] Lightner DA, Gawronski JK, Bouman TD. The octant rule. 7. Deuterium as an octant perturber. J Am Chem Soc, 1980, 102: 1983-1990.
[20] Albinsson B, Norden B. Excited-state properties of the indole chromophore: electronic transition moment directions from linear dichroism measurements: effect of methyl and methoxy substituents. J Phys Chem, 1992,96: 6204-6212.
杂志排行

Asian Journal of Traditional Medicines的其它文章
- Exploring the active ingredients, potential targets and pathways of quassinoids in Simaroubaceae plants by network pharmacology approachs
- Main chemical constituents and pharmacological properties of Harrisonia perforata (Blanco) Merr.
- Chemical constituents of the genus Pithecellobium:a systematic review
- Chemical constituents and pharmacological effects of the fruits of Camptotheca acuminata: a review of its phytochemistry
- Studies on the Chemical Components and Biological Activities of Stellera chamaejasme L.