二氢乳清酸脱氢酶突变对米勒综合征患者体内蛋白及功能的研究*
2018-03-29方静娴梁建英
方静娴,陈 蕾,梁建英,钱 虹,唐 翔
(1.南方医科大学口腔医院/广东省口腔医院儿童口腔科,广州 510280;2.南方医科大学口腔医院/广东省口腔医院口腔综合科,广州 510280;3.广州医科大学附属广州市妇女儿童医疗中心妇科,广州 510260)
线粒体是双膜封闭器官,因其能量转化功能被认为是真核细胞的能量加工厂。此外,线粒体也参与到钙和离子储存以及信号传导、细胞分化、细胞凋亡、细胞周期调控和细胞生长等重要功能之中[1]。近来有研究表明,线粒体功能异常可引起一系列疾病,而线粒体功能状态也与老龄化等问题可能存在关联[2-3]。考虑到腺苷三磷酸(ATP)的产生主要依赖于氧化呼吸链的作用,线粒体遗传物质的维护对于个体维持健康状态是相当重要的[4-5]。
核酸嘧啶的合成通过两条途径:从头嘧啶合成途径和补救途径。二氢乳清酸脱氢酶(DHODH)催化从头嘧啶合成途径第四步反应,将二氢乳清酸(DHO)转化成为乳清酸盐[6-7]。DHODH是整个核酸嘧啶合成中惟一存在于线粒体内膜的限速酶,而其他的限速酶均存在于细胞质中。DHODH催化DHO氧化为乳清酸盐,将电子传递给铝氧化呼吸分子辅酶Q,通过结合酶还原共同体黄素单核苷酸[8]。因此,DHODH依赖于辅酶Q,进而将线粒体氧化呼吸链和核酸嘧啶生物合成从功能上联系起来。
DHODH有两个结合位点。其底物DHO结合于第一位点,并通过共同底物电子受体而被氧化。形成乳清酸盐后,辅酶结合于其第二位点,并从共同底物处接收一个电子。经DHODH合成的乳清酸盐在尿苷-磷酸合酶(UMPS)复合体的作用下接着转化为尿苷酸(UMP),此即UMP合成[9-10]。
米勒综合征是轴后面骨发育不全,通常也以Wildervanck-Smith综合征为人所知。其临床症状包括严重小下颌、唇裂和(或)腭裂,轴背部位如四肢发育不全或发育缺陷,眼睑缺损以及多生乳头[11-12]。近来发现米勒综合征的致病基因是DHODH,其定位于染色体16q22[13]。从外显子2到外显子9,共发现13个突变体[13-14]。然而这些突变体是如何导致米勒综合征的发生却尚待研究。
小鼠体内,通过运用DHODH抑制剂来氟米特(Leflunomide/LFN)作用于怀孕小鼠,可引起一系列四肢及颌面发育缺陷,最主要的共同表现包括露脑畸形、腭裂和眼睑无法闭合[15]。因此,DHODH突变引起米勒综合征的发现揭示了DHODH在颌面和四肢发育过程中尚待发现的新作用。本研究中,观察3种与米勒综合征相关DHODH突变体在线粒体中的蛋白稳定性、定位和DHO-依赖酶活性,以期了解DHODH在米勒综合征中发挥的作用。
1 材料与方法
1.1抗体和试剂 抗-DHODH、抗HA和抗TFAM(线粒体转录因子A)抗体由九州大学临床检查部分子检查室提供。抗BAP37购于美国SantaCruz生物科技公司。抗-β-actin抗体购于美国Sigma公司。MitoTrackerRed购于美国Invitrogen公司。L-DHO购于美国Sigma-Aldrich公司。
1.2方法
1.2.1细胞培养 人宫颈癌HeLa细胞培养于DMEM培养液(美国Sigma-Aldrich)以及10%牛胚胎失活血清(FBS)。细胞系维持培养于37 ℃含5% CO2的培养箱中。
1.2.2表达载体 DHODH cDNA表达构建采用标准方法。野生和突变型DHODH cDNA通过BamHⅠ/XhoⅠ酶切位点加入载体pcDNA(Invitrogen)。DHODH cDNA包括推测的第一个甲硫氨酸位点通过人宫颈癌HeLa细胞的cDNA文库得以扩增,通过配对引物,即:5′-CAG AGT CTT CTG CCT CCC TG-3′和5′-AGG CAG AAG ACT CTG-3′。然后BamHⅠ和XhoⅠ位点分别通过二次PCR加入到5′和3′端,通过引物5′-GCG TGG AGA CAC CTG AAA AAG C-3′和5-CGA GTC ACC TCC GAT GAT CTG CTC C-3′。PCR产物经BamHⅠ和XhoⅠ酶切。pcDNA5/FRT载体(美国Invitrogen)经BamHⅠ和XhoⅠ酶切后的片段同编码HA标记的DNA片段结合在一起,形成编码DHODH的DNA片段,该载体命名为pDHODH-HA。
1.2.3构建突变DHODH蛋白质粒 DHODH突变体G202、R346W和R135C由pDHODH-HA产生,突变由PCR位点导向诱变产生[16]。所有PCR产生片段经插入至pGEMT载体(美国Promega)后测序确认,表达载体与pcDNA5/FRT共同构建。
为构建可稳定表达细胞系,按照生产商说明书HeLa Flp-InTMT-RexTMcells(美国Invitrogen)经LipofectamineTM2000 (Invitrogen)共转染于包含DHODH 和 pOG44的pcDNA5/FRT/TO。转染后,细胞培养于含有潮霉素B (200 μg/mL)和灭瘟素S (10 μg/mL,美国Invitrogen)的DMEM培养液。选择培养基培养3~4周后,挑选独立菌株,转移培养于独立培养皿,并用抗HA抗体检测表达。当加入DOX(doxycycline;1 μg/mL)于培养液时,可在指定时期诱导相应蛋白的表达。
1.2.4蛋白免疫印记检测 HeLa细胞裂解于TNE裂解液(50 mmol/L Tris/HCl,pH 7.5,1 mmol/L EDTA,150 mmol/L NaCl和0.5% Nonidet P40)。将20 μg提取蛋白上样于十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)电泳分离,并用下文所提及的具体抗体进行免疫印迹检测。检测信号通过辣根过氧化物酶(HRP)-标记抗兔免疫球蛋白G(IgG)和增强化学发光(ECL)试剂(GE Healthcare)显影。化学发光结果通过预冷电荷耦合设置(CCD)相机(LAS1000plus;Fuji Photo Film)进行记录和定量分析。
1.2.5亚线粒体定位 细胞收集器将HeLa细胞(1×108个)收集后悬浮于PBS溶液,经离心后沉淀,并用同质化缓存液清洗。上清液以10 000×g离心6 min后,将沉淀物作为原始线粒体部分。取20 μg线粒体处理于低渗液(10 mmol/L Hepes/KOH,pH 7.4 和1 mmol/L EDTA)于 4 ℃裂解线粒体外膜。经过低渗处理后,10 000×g离心6 min以获得纯化线粒体。将该线粒体组分悬浮于低渗缓冲液并用200 μg/mL蛋白酶K(含或不含1% Triton X-100)于冰上消化20 min。加入15% 三氯乙酸沉淀蛋白。沉淀溶于样品缓冲液(6% SDS,150 mmol/L氨基甲烷,10 mmol/L EDTA 和 25%甘油后经SDS/SPS-PAGE电泳和印迹分析检测。
1.2.6HeLa细胞免疫荧光成像 HeLa细胞经含500 nmol/L MitoTracker®Red (Invitrogen)处理20 min(按生产厂商说明书)。细胞固定渗透处理后,处理于1∶200稀释含抗HA和抗DHODH血清的1% BSA/PBS溶液1 h。处理后的样本置于Superfrost(Matsunami)。荧光图像经荧光显微镜(Biorevo,KEYENCE)获取分析。
1.2.7氧化呼吸链活性生化检测 分光光度法(U-3210,日立)检测DHODH依赖氧化呼吸酶活性。稳定表达DHODH的HeLa细胞冰浴15 min裂解于低渗缓冲液(2.5 mmol/L Tris/HCl,pH 7.5 和 2.5 mmol/L MgCl2),然后超声裂解15 s,样板用于检测氧化呼吸链复合物活性。
标准反应缓冲液包含50 mmol/L磷酸钾,5 mg/mL BSA和2.5 mmol/L MgCl2。检测不同复合体用不同的反应底物(电子供体及电子受体),每组反应都由受检测的特殊氧化呼吸链复合体的对应抑制剂来终止。每组参与反应的蛋白浓度通过二喹啉甲酸(BCA)蛋白检测法(Thermo Scientific)确定。每组复合体的活力通过每分钟每毫克底物氧化或还原率的改变来计算。检测每组复合体活力时加入其余氧化还原复合体的抑制剂,以确保所检测活力仅反映所检测酶复合体活力。每组数据检测于同一日,在相同条件下,重复3次。
1.2.7.1DHO-泛琨氧化还原酶活性 DHODH活力在37 ℃时通过分光光度法检测600 nm还原2,6-二氯靛酚(2,6-dichlorophenol-indophenol,DCPIP;用作虚拟电子受体)吸光度的降低程度。总的说来,反应由1 mL标准反应缓存液中的20 mmol/L DHO和50 μmol/L DCPIP启动,并包含2 μg的rotenone(复合体Ⅰ抑制剂),2 μg的抗霉素A(复合体Ⅲ抑制剂),5 mmol/L的NaN3(复合体Ⅳ抑制剂)及0.1 mg全细胞溶液。检测结果显示为nmol/μg蛋白。反应体系通过加入2 μg的来氟米特(leflunomide,LFN)终止。
1.2.7.2琥珀酸脱氢酶(氧化体呼吸链复合体Ⅱ)活力 琥珀酸脱氢酶(复合体Ⅱ)活力通过分光光度法检测于37 ℃。反应由含50 μmol/L DCPIP和20 mmol/L琥珀酸,2 μg的rotenone,2 μg的抗霉素A,5 mmol/L NaN3和0.1 mg全细胞液的1 mL标准反应缓存液发起。检测结果显示为nmol/μg蛋白。
1.2.7.3细胞色素C还原酶(复合体Ⅲ)活性 细胞色素C还原酶(复合体Ⅲ)活力通过分光光度法于37 ℃检测。通过检测细胞色素C于550 nm吸光度的降低程度来检测。反应由含5 mmol/L癸基泛醌和2 μg的鱼藤酮,5 mmol/L的NaN3,60 μmol/L细胞色素C和0.1 mg全细胞液的1 mL标准反应缓存液发起。反应通过加入2 μg的抗霉素A终止,检测结果显示为nmol/μg蛋白。
1.2.7.4细胞色素酶C氧化还原酶活性 DHO:细胞色素酶C氧化还原酶活性评估方法与1.2.7.3细胞色素酶C还原酶活性评估方法相似。检测环境与条件也与复合体Ⅲ相近,但将20 mmol/L DHO作为供体底物,并通过加入2 μg的抗霉素A终止反应。
1.2.8蛋白稳定性分析 野生型和突变型DHODH-HA经DOX诱导24 h产生后,分别在次日0、2、4、8 h后加入50 μg/mL放线菌酮(CHX),并收集细胞。提取总蛋白以进行蛋白免疫印迹检测,使用多克隆抗DHODH和抗HA抗体。检测分析条带信号。

2 结 果
2.1建立稳定表达DHODH蛋白的HeLa细胞系 构建载体转入细胞后,建立了在DOX诱导下可稳定表达包含DHODH-HA组合蛋白的细胞系(DHODH蛋白C末端包含HA标记)。经DOX诱导24 h后,同非诱导细胞系相比,组合DHODH-HA的表达量翻了5倍(图1A)。DHODH-HA蛋白经HA抗体染色后诱导效果(图1B)及诱导前未经HA抗体染色的蛋白表达情况显示内、外源性DHODH均准确定位于线粒体。

A:HA标记野生型DHODH蛋白印迹结果;B:融合DHODH蛋白细胞免疫荧光染色结果
图1野生型DHODH蛋白表达和定位
2.2米勒综合征相关联DHODH-HA的表达和定位 为观察米勒综合征中发现的突变蛋白的处理、定位和功能,将G152R、R346W和R135C突变引入DHODH cDNA(图2A)。经DOX诱导24 h后,同非诱导细胞系相比,组合DHODH-HA的表达量翻了5倍(图2B)。经免疫荧光染色检测,R135C-突变型DHODH显示了正常的细胞内定位。抗HA抗体显示其存在于HeLa细胞中的线粒体内(图2C)。而G202A和R346W突变型则显示弱染色,虽然同样定位于线粒体内。R346W型构建体显示在线粒体外有弱溶解(图2C)。
2.3突变DHODH蛋白在线粒体内的定位 经低渗处理后,野生型和所有的DHODH突变蛋白均可被蛋白酶k剪切消化(图2D,第4列)。当线粒体内膜经非离子洗涤剂TritonX-100处理后,TFAM和DHODH都被蛋白酶k消化(图2D,第5列)。实验中构建的3种突变DHODH蛋白均准确存在于线粒体内膜空间内或内膜内(图2D,第2、3、4行)。
A:DHODH及米勒综合征相关突变结构图示;B:HA标记野生型DHODH蛋白诱导后的表达情况;C:融合突变型DHODH免疫荧光染色结果;D:外源性野生型和突变型DHODH蛋白在线粒体内的亚定位;1:完整线粒体;2.线粒体外膜经蛋白酶处理后;3、4:线粒体外膜经低渗法裂解后;5:线粒体经裂解及蛋白酶处理后
图2米勒综合征相关突变蛋白的表达和定位
2.4R346W和G202A突变DHODH的蛋白稳定性减低 经DOX诱导后用CHX抑制新蛋白的表达。DHODH蛋白总量经CHX处理0、2、4、8 h后收集后检测。野生型和R135C型DHODH蛋白水平8 h处理后相对一致(图3),突变蛋白自身降解加快。而在实验中,并未观察到β-actin的不稳定性(图3A)。
2.5突变DHODH蛋白的DHO依赖泛醌和细胞色素C氧化还原酶活性 通过检测野生型和两株可稳定表达突变型R135C DHODH的细胞检测氧化呼吸复合体的酶活性,发现在HeLa细胞转染体中,诱导后的野生型DHODH蛋白的DHO依赖酶活性较突变型增加了5倍;而在突变型R135C中,诱导后相应蛋白表达量增加了5倍,但其DHO依赖酶活性却没有明显的变化(图1A和图4A)。经DOX诱导后,G202A和R346W型DHODH的DHO依赖酶活性增加了2倍,与其蛋白表达量的增加幅度相对应,即G202A和R346W型DHODH蛋白的功能酶活性存在,而其蛋白稳定性减低(图4B)。而R135C和野生型DHODH两组中复合体Ⅱ、复合体Ⅲ和复合体Ⅱ~Ⅲ联合体的多项都没有差别(图4C)。
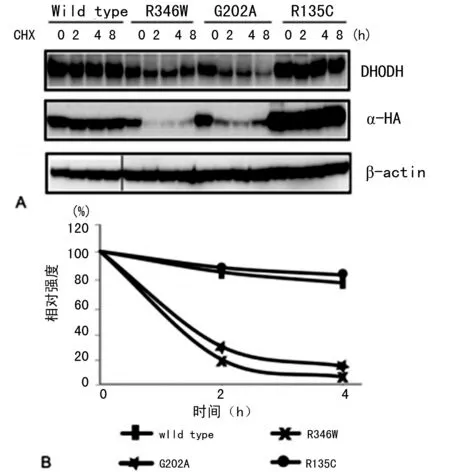
A:经CHX处理0、2、4、8 h后野生型和突变型DHODH蛋白表达情况;B:图A相关图像资料量化结果
图3突变型DHODH蛋白稳定性检测结果

A:R135C型DHODH转染细胞中降低的DHO依赖酶活性;B:表达G202C和R346W型DHODH蛋白细胞中DHO依赖还原酶活性检测;C:表达R135C和野生型DHODH的细胞中线粒体复合体Ⅱ和Ⅲ活性检测结果。a:P<0.05,与对照组比较
图4米勒综合征相关DHODH突变蛋白酶活性检测
3 讨 论
米勒综合征以常染色体隐性异常或复合杂合子模式遗传[13]。米勒综合征阻断了第一和第二鳃弓的发育。DHODH基因突变时如何导致鳃弓的异常发育目前尚不清楚。但需要指出的是DHODH是从头嘧啶合成6个限速酶中的第4个[8],该途径中的其余5个酶分别组成两个不同的酶复合体,并均存在于细胞质中。前3个酶形成了多重酶复合体CAD(氨基甲酰磷酸合成酶、天冬氨酸转氨基甲酰酶和二氢乳清酸酶),而第5和第6个酶形成了UMPS。
UMPS基因编码具有乳清酸磷酸核糖转移酶和乳清苷酸脱羧酶双重功能的酶[17]。UMPS基因复合杂合子突变引起乳清酸尿症,一种较少见的常染色体隐性遗传。其临床表现包括了巨幼红细胞性贫血症和乳清酸结晶尿,并常伴有智力和身体发育上的迟缓[18]。然而,作为UMPS突变并不会引起与米勒综合征相似的发育异常。这提示,米勒综合征中的嘧啶或嘌呤缺乏并不是导致颌面发育异常的原因。
WHITE等[19]曾报道LFN(一种DHODH抑制剂),可引起斑马鱼神经嵴发育的完全停止以及哺乳动物神经嵴干细胞自我更新的减缓。LFN发挥这些作用是通过抑制神经嵴发育所需基因的转录延长来实现的[17]。神经嵴细胞的特殊发育途径对黑素瘤的形成有直接关系,提示了DHODH也参与了神经嵴细胞的增殖和发育。
甲氨蝶呤是一种从头嘌呤生物合成的抑制剂,而其抗增殖作用通常被认为是由其对二氢叶酸还原酶(DHFR)的抑制作用而产生的。婴幼儿中甲氨蝶呤最常导致的发育异常胚胎疾病包括:发育缺陷、颅缝早闭、小颌以及骨骼异常,而这些症状与米勒综合征个体所表现的个体症状相类似[20]。然而,DHFR自身突变并不会导致颌面异常。考虑到米勒综合征部分程度上是由于线粒体功能异常,甲氨蝶呤有可能不仅仅作用于DHFR,也作用于神经嵴细胞的线粒体。
许多出生缺陷,例如米勒综合征和甲氨蝶呤所致胚胎病,都与颌面发育异常相关。这些数据说明神经嵴细胞的发育涉及颌面发育[23]。而米勒综合征患者所表现临床症状有可能是由于DHODH功能缺失所引起的神经嵴细胞发育缺陷所致。
人体内DHODH蛋白主要由两部分组成:较大的C末端(蛋氨酸78-精氨酸396)和较小的N末端(蛋氨酸30-亮氨酸68),其间通过一个扩展环连接。较大C末端域被描述为α/β-筒折叠,其中央筒有8个平行β链,外围环绕着8个ɑ螺旋。此结构中存在一些近端还原位点,并以部分带点或复极化链收尾(Gln47、Tyr356、Thr360和Arg135)。从人类到恶性疟原虫精氨酸135均存在于泛醌结合位点,其均表现出了保守性。本研究观察了DHODH突变型R135C,该型在米勒综合征患者中也被监测出。该突变型具有准确的线粒体定位功能,但却缺乏DHO依赖酶活性。由于DHODH的α-和β-筒域形成了酶活性反应的作用位点,与α-和β-筒域相互作用的化合物可阻断DHODH活性。G202A和R346W存在于DHODH的ɑ-筒域内;G202A和R346W突变体可能是由于蛋白质构象上的改变而导致蛋白质稳定性降低。
本研究涉及实验中,MitoTracker®Red染色的细胞与HA染色的细胞部分重叠,提示细胞中表达的野生型DHODH-HA蛋白准确表达于线粒体中。相同的染色结果可观察于经抗DHODH抗体处理的染色结果中(图1B),提示内、外源性DHODH均准确定位于线粒体。实验中用同样方法成功构建了可稳定表达突变型DHODH的细胞株(图2),对突变型蛋白的定位研究结果提示突变型蛋白都主要存在于线粒体内,尽管一小部分R346W存在于细胞质内。为检测野生型和突变型DHODH-HA蛋白在线粒体内的定位,通过低渗法裂解线粒体外膜后(图2D,每组中第3、4列),可观察到BAP37(一种线粒体内膜蛋白),可被蛋白酶k消化;而TFAM(存在于线粒体基质内的蛋白),则因受到内膜保护而不被蛋白酶k消化(图2D,第4列较低行),从而说明低渗法可保持线粒体内膜的完整性。通过低渗法完整提出线粒体对判断突变型DHODH的定位提供了保证。
为检测突变DHODH蛋白减少是否是由于蛋白稳定性降低所致,本研究培养了可稳定表达野生型或突变型DHODH蛋白的HeLa细胞,并在经DOX诱导后用CHX抑制新蛋白的表达。DHODH蛋白总量经CHX处理0、2、4、8 h后收集后检测。野生型和R135C型DHODH蛋白水平8 h处理后相对一致(图3),突变蛋白自身降解加快。在实验中,并未观察到β-actin的不稳定性(图3A)。由于这些突变都存在于DHODH蛋白的α螺旋中,也有可能是突变导致了蛋白结果稳定性的降低从而加速了蛋白的降解。R346W和G202A突变导致的DHODH蛋白稳定性降低,从某种程度上对米勒综合征的发生发挥了作用。
由于R135C型DHODH突变蛋白并未显示明显的稳定性或内膜空间定位功能异常,实验接着检测了该突变是否影响酶活性。结果显示突变型R135C DHODH缺失其供电子功能。有研究显示,Arg135残基存在于DHODH蛋白的泛醌结合位点上,提示了R135C突变型有可能无法结合于泛醌发挥作用。
考虑R135C突变是否会影响其他氧化呼吸复合体的功能,研究中同时也比较了DOX诱导后的酶活性,结果显示无差别。提示R135C DHODH并没有直接影响其他的氧化呼吸复合体。
本研究观察到,G202A和R346W突变型引起了蛋白稳定性的缺失和R135C突变型底物诱导酶活性的受损,这些提示DHODH蛋白活性受损与米勒综合征表现型相关。
[1]MCBRIDE H M,NEUSPIEL M,WASIAK S.Mitochondria:more than just a powerhouse[J].Curr Biol,2006,16(14):R551-560.
[2]DIMAURO S,SCHON E A.Mitochondrial disorders in the nervous system[J].Annu Rev Neurosci,2008,31:91-123.
[3]FRENZEL M,ROMMELSPACHER H,SUGAWA M D,et al.Ageing alters the supramolecular architecture of OxPhos complexes in rat brain cortex[J].Exp Gerontol,2010,45(7-8):563-572.
[4]WALLACE DC.Mitochondrial DNA mutations in disease and aging[J].Environ Mol Mutagen,2010,51(5):440-450.
[5]TUPPEN H A,BLAKELY E L,TURNBULL D M.Taylor RW.Mitochondrial DNA mutations and human disease[J].Biochim Biophys Acta,2010,1797(2):113-128.
[6]LOFFLER M,GREIN K,KNECHT W,et al.Dihydroorotate dehydrogenase Profile of a novel target for antiproliferative and immunosuppressive drugs[J].Adv Exp Med Biol,1998,431:507-513.
[7]EVANS DR,GUY HI.Mammalian pyrimidine biosynthesis:fresh insights into an ancient pathway[J].J Biol Chem,2004,279(32):33035-33038.
[8]RAWLS J,KNECHT W,DIEKERT K,et al.Requirements for the mitochondrial import and localization of dihydroorotate dehydrogenase[J].Eur J Biochem,2000,267(7):2079-2087.
[9]LOFFLER M,JOCKEL J,SCHUSTER G,et al.Dihydroorotat-ubiquinone oxidoreductase links mitochondria in the biosynthesis of pyrimidine nucleotides[J].Mol Cell Biochem,1997,174(1-2):125-129.
[10]WALSE B,DUFE VT,SVENSSON B,et al.The structures of human dihydroorotate dehydrogenase with and without inhibitor reveal conformational flexibility in the inhibitor and substrate binding sites.[J].Biochemistry,2008,47(34):8929-8936.
[11]MILLER MF,FINEMAN R,SMITH DW.Postaxial acrofacial dysostosis syndrome[J].J Pediatr,1979,95(6):970-975.
[12]DONNAI D,HUGHES HE,WINTER RM.Postaxial acrofacial dysostosis (Miller) syndrome[J].J Med Genet,1987,24:422-425.
[13]NG SB,BIGHAM AW,BUCKINGHAM KJ,et al.Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome[J].Nat Genet,2010,42(9):790-793.
[14]KINOSHITA F,KONDOH T,KOMORI K,et al.Miller syndrome with novel dihydroorotate dehydrogenase gene mutations[J].Pediatr Int,2011,53(4):587-591.
[15]FUKUSHIMA R,KANAMORI S,HIRASHIBA M,et al.Teratogenicity study of the dihydroorotate-dehydrogenase inhibitor and protein tyrosine kinase inhibitor Leflunomide in mice[J].Reprod Toxicol,2007,24(3-4):310-316.
[16]UCHIUMI T,OHGAKI K,YAGI M,et al.ERAL1 is associated with mitochondrial ribosome and elimination of ERAL1 leads to mitochondrial dysfunction and growth retardation[J].Nucleic Acids Res,2010 (38):5554-5568.
[17]MCCLARD RW,BLACK MJ,LIVINGSTONE LR,et al.Isolation and initial characterization of the single polypeptide that synthesizes uridine 5-monophosphate from orotate in Ehrlich ascites carcinoma.Purification by tandem affinity chromatography of uridine-5-monophosphate synthase[J].Biochemistry,1980,19(20):4699-4706.
[18]SUCHI M,MIZUNO H,KAWAI Y,et al.Molecular cloning of the human UMP synthase gene and characterization of point mutations in two hereditary orotic aciduria families[J].Am J Hum Genet,1997,60(3):525-539.
[19]WHITE RM,CECH J,RATANASIRINTRAWOOT S,et al.DHODH modulates transcriptional elongation in the neural crest and melanoma[J].Nature,2011,471(7399):518-522.
[20]RUCKEMANN K,FAIRBANKS LD,CARREY EA,et al.Leflunomide inhibits pyrimidine de novo synthesis in mitogen-stimulated T-lymphocytes from healthy humans[J].J Biol Chem,1998,273(34):21682-21691.
[21]MILUNSKY A,GRAEF JW,GAYNOR MF JR.Methotrexateinduced congenital malformations[J].J Pediatr,1968,72(6):790-795.
