医学信息学在罕见病诊疗中的研究进展及应用
2018-03-28姜召芸卢宇蓝弓孟春史文钊张抒扬周文浩
姜召芸,卢宇蓝,余 乐,弓孟春,史文钊,张抒扬,周文浩
1神州数码医疗科技股份有限公司,北京 100080 2复旦大学附属儿科医院 儿科研究所,上海 201102 3复旦大学儿童发育与疾病转化医学中心,上海 201102 4上海市出生缺陷重点实验室,上海 201102 5中国医学科学院罕见病研究中心,北京 100730 6中国医学科学院 北京协和医学院 北京协和医院,北京 100730
1 概述
目前国际上尚无对罕见病的统一定义。在美国,罕见病被定义为影响少于20万美国人的疾病;在欧盟,罕见病被定义为发病率在万分之五以下的疾病[1]。目前公认的罕见病数量为6000~7000种[1]。其中近75%为儿童罕见病,约5000种,影响全世界数百万儿童[2]。一项对欧洲17个国家8种罕见病的调查研究显示,25%的患者需要等待5~30年才被确诊,40%的患者最初诊断不正确,部分患者未被诊断[3]。尽管这些疾病通常是慢性和进行性的,但若能早期诊断(如新生儿筛查),并通过标准和/或靶向治疗进行最佳管理,则长期并发症可被减轻或延迟[4]。
大多数罕见病由单基因突变引起,这些罕见的遗传性疾病也被称为孟德尔遗传病或单基因疾病。因此,罕见病是很好的病因研究模型,可为很多常见疾病的发病机制研究提供重要线索。随着新兴技术尤其是基因组学分析和相关序列分析技术的兴起,以及个性化医疗的出现,可发现更多致病基因,增加疾病的亚型数量,进一步促进罕见病研究方法的发展[1]。
医学信息学是新兴交叉学科,研究者为追求科学探究、问题解决和决策制定,对生物医学数据、信息、知识进行有效利用,以提高人类健康[5]。医学信息学研究的最新进展,包括电子病历、生物信息技术、医院信息系统、决策支持系统、影像信息技术、远程医疗与互联网以及数据标准等。罕见病作为病因研究模型,有利于研究整合不同来源数据,这些来源不同的数据包括临床和生理数据(如肌张力检查和肌酸激酶浓度)、分子数据(如基因表达和基因型)、生物样本库数据和模式生物/疾病模型信息[6]。对罕见病不同维度的数据进行研究会促进医学信息学技术的发展。本文将详细阐述医学信息学的关键技术及其在罕见病诊疗中的应用。
2 医学信息学关键技术
2.1 罕见病临床术语标准化
罕见病种类繁多且表型复杂多样,不仅在不同疾病间存在差异,同一疾病的不同患者在表型上也可能不同,具有高度的临床异质性,为罕见病数据整合带来很大挑战[7]。目前国际上已形成一批广泛认可、应用效果良好的医学语义标准,如国际卫生术语标准制定组织研发的SNOMED-CT(systematized nomencl-ature of medicine-clinical terms)[1]、美国国立图书馆研发的统一医学语言系统UMLS(unified medical language system)[8]、德国柏林夏里特医学院研发的人类表型本体(the human phenotype ontology,HPO)[9]等,这些本体系统在规范描述不同类别的临床数据方面发挥重要作用。其中SNOMED-CT是世界上最全面、多语言的临床医学术语产品,包含大多数方面的临床信息,如人体结构、临床发现、临床操作、事件、药物等19个临床领域,超过50个国家在使用。但SNOMED-CT目前缺少中文版,在中文电子病历中的应用受限。UMLS包括超级叙词表(metathesaurus)、语义网络(semantic network)、情报源图谱(information sources map)和专家词典(SPECIALIST lexicon)4部分。其中超级叙词表是生物医学概念、术语、词汇及其涵义、等级范畴的广泛集成。UMLS的语义网络不仅运用了常规语义控制手段,如语义等级、属分、相关关系控制,且在语义规范和语义关系分析、延伸等多方面有许多创新。HPO解决了对罕见病表型标准化词汇表的需求[10],在数项大型国际项目中用来对患者进行编码。HPO是一个有向无环图,表示超过10 000个表型异常,其中节点(HPO项)通过“is a”关系彼此连接。 HPO由诸多医学领域专家创建,以适应不同来源表型数据的编码,如实验室试验、图像、图表和临床解释[11]。由欧洲生物信息研究所(the European Bioinformatics Institute,EMBL-EBI)、葛兰素史克(Glaxo Smith Kline, GSK)和威康信托桑格研究所(the Wellcome Trust Sanger Institute, WTSI)合作开发的基于基因组实验的药物靶点证据知识库CTTV(the center for therapeutic target validation),将生成的数据与EMBL-EBI、GSK和WTSI 资源中的现有数据进行整合,使用EFO(experimental factor ontology)作为其应用本体。EFO为其他本体提供整合框架,例如ORDO、ChEBI,the gene ontology 和Uberon[12]。
建立统一的罕见病语义网络,规范疾病症状描述,对罕见病研究至关重要。 构建语义网络的基础是对杂乱无章的非结构化临床文本信息结构化处理。临床自然语言处理(nature language processing,NLP)利用多层算法融合,对文本信息进行分词、逻辑关系处理、特征提取,以完成对非结构化文本信息的处理。临床NLP为临床医生提供关键的患者病例详细信息,这些详细信息通常被锁定在非结构化临床文本中,并散布在患者的健康记录中。语义分析是临床NLP研究的主要目标之一,通过鉴定临床实体(如患者、临床医生)和事件(如疾病、治疗)并呈现其关系来解锁文本的意义[13]。语义互操作性是实现语义网络的关键,通常以本体和可控词汇的形式存在。本体是“概念化的明确说明”,是一种以结构化的方式描述领域知识的方法[14]。如果以一个本体作为参考(如the gene ontology和SNOMED-CT),那么其对真实世界数据的表示则成为个标准,数据整合也会变得容易[15]。
2.2 影像组学数据分析
影像组学被定义为数量映射,即对大量医学图片特征进行提取、分析和建模[16]。影像组学的研究可分为数据选择、医学成像、特征提取、探索分析和构建模型5个阶段:(1)数据选择:影像组学的分析始于成像步骤、感兴趣体积(volume of interest, VOI)和预期靶点的选择。(2)医学成像:VOI可被手动或半自动化切割[17]。切割决定图像中被分析元素,切割的多样性在影像特征的评价中引入了偏差。(3)特征提取:影像组学的本质是定量图像特征的高通量提取以表征VOI。特征值依赖于图像预处理(如滤波或强度离散化)和重构(如滤波反投影或迭代重建)等因素。此外,在特征命名、数学定义、方法论和特征提取算法的软件实现中存在差异[18]。(4)探索分析:影像特征和非影像特征可与预测目标相结合以创建单个数据集。高度相关的影像特征组可以通过聚类来识别,而这些特征可以简化为每个聚类的单一原型特征。从多重分割、多次成像和幻影研究中收集的辅助特征数据可以用来评估特征的鲁棒性。(5)构建模型:影像组学建模涉及3个主要方面,即特征选择、建模方法学和验证。特征选择应该是数据驱动的,因为可能的影像特征范围广,这种分析应该以强有力和透明的方式进行。关于建模方法的选择,确定最佳的机器学习方法是迈向临床决策支持的关键一步。因此,在理想的情况下,应该采用多种机器学习方法[19],全面记录实施情况。未经验证的模型价值有限,验证也是完整的影像分析不可缺少的组成部分。
影像组学在罕见病应用中很重要的一个方面是患者人脸识别功能。Face2Gene通过对来自真实患者的数据、数以百万计的数据点进行训练,结合基因组学,通过患者面部特征对疾病进行诊断。将面部识别软件与临床知识(特征注释和人体测量)相结合,从二维面部照片中能够检测畸形特征和可识别的人类畸形模式。所进行的测量形成脸部描述符,可与其他描述符进行比较,并以图形方式显示为面具[20]。
虽然目前影像组学处理流程已经比较完善,但很多流程的优化仍是难题,如其中关键的分割算法的改进仍是挑战性问题,人工分割耗时耗力,自动分割鲁棒性和精度难以保证。随着近期深度学习浪潮的推动,基于深度机器学习的分析预测方法将是影像学未来发展方向之一,为预测准确率的提高提供了突破方向。另一方面,由于目前医院患者人数众多,影像检查费时费力,影像组学也应尽可能与临床特征相融合,成为临床医师更加信赖和认可的方法,从真正意义上发展为一种辅助诊断工具,提供更为便捷和放心的服务。
2.3 基因组学数据分析
目前基因组学分析已广泛应用于罕见病的研究。基因组及相关技术近几年在医学领域的应用发展迅速。随着全基因组、全外显子组测序价格的下降、计算速度的加快以及精度的提高,科学家们已经能够在单核苷酸分辨率的水平上测序整个基因组[21]。基因组学的大规模分析使单基因遗传病的致病病因可在分子水平或细胞水平进行定义,治疗也更加个性化和有针对性。
用于罕见病研究的基因组检测方法主要有3种:(1)基因panel:对相似表型相关的若干个基因的编码序列进行平行测序;(2)全外显子组测序(whole exome sequencing,WES):对人类基因组的所有已知编码区进行测序;(3)全基因组测序(whole genome sequencing,WGS):对整个人类基因组进行测序。 WES近年来主导了罕见病研究,外显子序列约占人类全部基因组序列的1%,却涵盖了大约85%的变异[22]。与WGS相比,WES在收集和分析基因组数据方面更为经济有效,基因panel测序提供了比WES更快的周转时间,但不能发现新的致病基因[23]。
2.4 表型组学和基因组学数据融合分析
表型组学是测量表型组的生物学领域,包括物理和生物化学特征,会随着基因突变和环境影响而发生改变[24]。表型信息对临床诊断决策至关重要,包括基因检测之前和基因检测之后。基因检测之前,表型信息可辅助锁定要研究的区域,帮助筛选检验项目。基因检测之后,表型信息可和基因信息关联起来对疾病进行解释。将表型信息和基因型信息结合起来进行研究对疾病的判别至关重要。
2.4.1 全基因组关联分析
全基因组关联分析(genome-wide association studies, GWAS)是研究某种基因变异与特定疾病表型之间关系的方法,属于正向遗传学研究范畴, 一般应用于检测单核苷酸多态性(single nucleotide polymorphism,SNP)和疾病之间的关系,如心脏病、糖尿病、自身免疫性疾病和精神疾病等[25]。针对群体中个体的表型(如是否患有某种疾病)进行研究,然后从成千上万种SNP中找出与该表型相关的基因型。若能从统计学上确认这些SNP与该表型之间具有联系,就可初步认定其是与该表型相关的基因型。随后,还需要在另一个种群中进行试验,验证上述发现是否正确。标准GWAS统计方法包括回归建模(线性和逻辑),并调整相关协变量,如性别、年龄、种族或民族。对于多重检验调整显著性阈值,普遍接受的全基因组显著性阈值为P<5.0×10-8[26]。GWAS已经走过10年的发展历史,数百个复杂性状的GWAS结果已被报道。目前,GWAS研究主要依赖于SNP 芯片,但是这些芯片无法覆盖所有的遗传变异,未来将以全基因组测序为基础。随着研究规模的扩大,发现的新致病位点对疾病的影响往往更小,对于发病率低的罕见病而言,研究受样本数量的限制,需提升技术来克服这些挑战[27]。
2.4.2 全表型组关联分析
全表型组关联分析(phenome-wide association studies,PheWAS)旨在研究与已知的基因变异相关联的表型,是与GWAS相反的研究方法,属于反向遗传学范畴。PheWAS方法已通过已知的基因型-表型关系得到验证,且使用PheWAS已能区分真正的多效性和临床共病,增加遗传发现的背景,还有助于定义疾病亚型。同时,PheWAS有利于研究数据收集,可能会改变药物用途。深入理解电子健康记录(electronic health record,EHR)数据,整合广泛且强大的表型数据, 将会创造丰富的资源,可以产生更多有效且详细的基因组-表型组分析,促进精准医学的新发现[26]。受具有多种表型队列的限制,PheWAS 2010年才被开发使用[28]。应用PheWAS算法分析7个已知关联的SNP-疾病对,其中4个被重复且P值在2.8×10-6和0.011之间。PheWAS算法还鉴定了19个之前未知的SNP与疾病在统计学上的关联(P<0.01)。
关于PheWAS的开放性的研究问题:是否能够从流行病学数据或通过重新利用收集到的EHR数据中全面捕捉到人类基因组的变异?考虑到不同表型组定义在特异性和粒度方面的差异,表征不同表型之间的表型相关性以及建立PheWAS特异性、显著性阈值成为目前研究热点[29]。
2.5 罕见病数据库/知识库
通过先进的医学信息学技术整合不同来源的数据,形成罕见病表型和遗传多样性之间的知识关联,建立相关数据库和知识库,对于罕见病诊疗至关重要。目前国际上已有很多数据库或知识库提供罕见病基因型与表型之间的关联:(1)OMIM(online mendelian inheritance in man)是一个综合且权威的关于人类基因与遗传变异的数据库[30],主要针对遗传性疾病,内容包括文本信息、相关参考信息、序列纪录、图谱和相关其他数据;(2)Orphanet是一个罕见病和治疗药物及其他资料的综合知识库,收录了近6000种罕见病各种相关信息,是目前世界上最权威和丰富的罕见病知识库[31];(3)DECIPHER(databasE of genomiC varIation and phenotype in humans using ensembl resources)是一个基于网络的可交互数据库,整合了一套旨在帮助解释基因组变异的工具,从患者遗传变异相关的各种生物信息学资源中检索信息以增强临床诊断[32];(4)GeneReviews主要由两大部分组成,即包含描述特定遗传疾病的标准化同行评议的论文和包含单个基因和表型信息的章节,目前共包含691个章节,在PubMed中可被索引;(5)ClinVar是一个公共数据库,其主要目的在于整合不同来源的数据,将基因变异位点、临床表型、实证数据以及功能注释等方面的信息,形成一个标准的、可信的、稳定的遗传变异-临床表型相关数据库,可免费访问[33];(6)HGMD(the human gene mutation database) 可提供人类遗传性疾病突变的综合性数据,已被广泛应用于临床,可实现针对单一突变的快速查询,同时支持各种先进的搜索应用[34];(7) GeneCards是人类基因综合数据库,提供基因组、蛋白质组、转录、遗传和功能上所有已知和预测的人类基因[35];(8)MalaCards是一个可搜索人类疾病及其注释的综合数据库,收录了约2万种疾病,在15个部分中描绘了广泛的注释主题,包括摘要、症状、解剖背景、药物、基因测试、变异和相关文献[36]。
对基因变异的最全面解读不仅需精心设计的数据库、分离分析和数据共享,还应进行适当的功能分析,如评估变异对剪接或基因表达的影响[37]。在当前基因组学时代,意义不明变异仍最常见[38]。
3 医学信息学技术在罕见病诊疗中的应用
3.1 罕见病致病基因的发现及诊断
罕见病由于其遗传性较好,可利用二代测序技术方便寻找致病基因[39]。WES在疾病致病基因的鉴定研究中取得了较大成果,不仅在单基因遗传病,还在多基因影响的复杂疾病中发现了大量相关基因。2010年,华盛顿大学孟德尔基因组学研究中心的研究小组发现了Miller综合征的遗传病因,其特征是严重小头畸形、唇裂和/或腭裂、四肢发育不良、眼睑和多余的乳头[40]。同一年,同样的方法成功鉴定了Kabuki(MLL2)[41],Schinzel-Giedion(SETBP1)[42]和Sensenbrenner(WDR35)[43]综合征的致病基因。2014年,加拿大FORGE(finding of rare disease genes)组织花费两年时间,汇集了加拿大各地21个遗传中心和3个科技创新中心的临床医生和科学家,对246例罕见病患者进行WES分析,发现了引发疾病的146个突变位点和67个异常基因[44]。
GWAS技术也被成功应用于罕见病致病基因的发现。GWAS已经鉴定了数千种与特定人类表型相关的常见遗传变异[45]。Duan等[46]利用GWAS分析方法对1492例烟雾病病例和5084例对照组进行研究,发现了烟雾病的新易感基因。同样,PheWAS在研究SNP-疾病对中也被成功应用,成为研究SNP-疾病对的关联性可用方法[28]。
3.2 罕见病药物的研发
因罕见病病因复杂、基因异质性高、病理机制未明、参与临床试验的患者数量少等问题,治疗罕见病的药物(也称孤儿药)在研发上具有很大的局限性。而医学信息学技术的发展对于突破这些局限性提供了有力帮助,在新药研发、老药新用等方面展示出良好的前景,如组学和信息学技术的发展为药物靶点发现提供了新契机;同时,临床记录与基因组学数据、药物间相互作用等多维度信息的整合分析,对安全、高效的老药新用策略具有重要意义。随着高通量技术的发展,人们对基因组的研究逐步深入,对罕见病发病机制的认识越来越清晰。一些用于治疗常见疾病的药物被发现可用于罕见病的治疗,如美国食品药品监督管理局(Food and Drug Administration,FDA)2009年批准将治疗勃起功能障碍的他达拉非(tadalafil)用于肺动脉高压的治疗[47]。FDA已批准约400种孤儿药,但这些孤儿药仅对5%的罕见病起作用,大多数罕见病的治疗仍依赖症状治疗甚至安慰剂治疗[48]。随着信息数据库和网络技术的发展,新的计算机模型将有助于加速罕见病临床前药物开发的目标识别和过程优化[49]。
3.3 罕见病注册登记平台的建立
在罕见病领域,注册登记系统被认为是开展临床研究的有力工具,有助于规划临床试验,改善患者的护理和医疗保健计划。数据质量被定义为数据集特征和特性的总和,这些数据集的特征和能力满足数据预期使用所产生的需求。在注册登记中,“产品”是数据,质量是指数据质量,意味着进入注册登记系统的数据已得到验证,可供分析和研究使用。通过对数据进行多维度的评估来确定数据的质量,包括评估数据的完整性、有效性、一致性、可比性、可访问性、实用性、及时性和预防重复记录[50]。医学信息学技术在罕见病注册登记平台中起到重要支撑作用。
4 展望
随着精准医学的发展,医学信息学关键技术(图1)在罕见病诊断、孤儿药开发等领域发挥巨大的推动作用。但这些技术的应用仍存在一定局限性,面临多种挑战。比如基因分析技术的发展,克服了罕见病致病基因未知、突变类型多变的困难,为罕见病能得到及时、准确的诊断创造了条件。但目前二代测序技术的应用还仅局限于大型医学或遗传学研究中心,且由于高通量测序数据的产生,数据保存和结果解读仍面临巨大挑战。
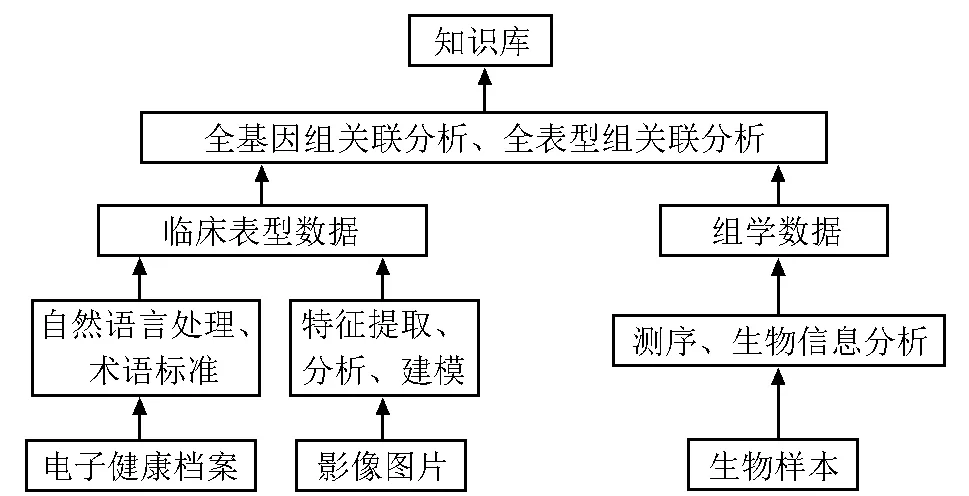
图 1 罕见病研究中的医学信息学技术路线
尽管目前存在各种挑战,但是精准医学的发展和医学信息学的不断进步,为罕见病的诊断和个性化治疗带来了前所未有的机遇。相信随着对罕见病研究的不断深入,医学信息学相关技术的不断发展,高通量测序等技术将在罕见病诊断和治疗中发挥越来越关键的作用。
[1] Fung KW, Richesson R, Bodenreider O. Coverage of rare disease names in standard terminologies and implications for patients, providers, and research [J]. AMIA Annu Symp Proc, 2014: 564- 572.
[2] Boycott KM, Dyment DA, Sawyer SL, et al. Identification of genes for childhood heritable diseases [J]. Annu Rev Med, 2014,65: 19- 31.
[3] Schieppati A, Henter JI, Daina E,et al. Why rare diseases are an important medical and social issue [J]. Lancet, 2008,371: 2039- 2041.
[4] Boycott KM, Rath A, Chong JX, et al. International cooperation to enable the diagnosis of all rare genetic diseases [J]. Am J Hum Genet, 2017,100: 695- 705.
[5] Liang J, Wei K, Meng Q, et al. Development of medical informatics in China over the past 30 years from a conference perspective and a Sino-American comparison [J]. Peer J, 2017, 5: e4082.
[6] Roos M, López Martin E,Wilkinson MD. Preparing data at the source to foster interoperability across rare disease resources [J]. Adv Exp Med Biol, 2017,1031: 165- 179.
[7] Ma’ayan A, Rouillard AD, Clark NR, et al. Lean big data integration in systems biology and systems pharmacology [J]. Trends Pharmacol Sci, 2014, 35: 450- 460.
[8] He Z, Chen Y, de Coronado S, et al. Topological-pattern-based recommendation of UMLS concepts for national cancer institute thesaurus [J]. AMIA Annu Symp Proc, 2016: 618- 627.
[9] Köhler S, Vasilevsky NA, Engelstad M, et al. The human phenotype ontology in 2017 [J]. Nucleic Acids Res, 2017, 45: D865-D876.
[10] Köhler S, Doelken SC, Mungall CJ, et al. The human phenotype ontology project: linking molecular biology and disease through phenotype data [J]. Nucleic Acids Res, 2014, 42: D966-D974.
[11] Greene D, BioResource N, Richardson S, et al. Phenotype similarity regression for identifying the genetic determinants of rare diseases [J]. Am J Hum Genet, 2016, 98: 490- 499.
[12] Sarntivijai S, Vasant D, Jupp S, et al. Linking rare and common disease: mapping clinical disease-phenotypes to ontologies in therapeutic target validation [J]. J Biomed Semantics, 2016, 7: 8.
[13] Velupillai S, Mowery D, South BR, et al. Recent advances in clinical natural language processing in support of semantic analysis [J]. Yearb Med Inform, 2015, 10: 183- 193.
[14] Machado CM, Rebholz-Schuhmann D, Freitas AT, et al. The semantic web in translational medicine: current applications and future directions [J]. Brief Bioinform, 2015, 16: 89- 103.
[15] De Silva TS, MacDonald D, Paterson G, et al. Systematized nomenclature of medicine clinical terms (SNOMED CT) to represent computed tomography procedures [J]. Comput Methods Programs Biomed, 2011, 101: 324- 329.
[16] Lambin P, Leijeaar RTH, Deist TM, et al. Radiomics: the bridge between medical imaging and personalized medicine [J]. Nat Rev Clin Oncol, 2017, 14: 749- 762.
[17] Polan DF, Brady SL,Kaufman RA.Tissue segmentation of computed tomography images using a random forest algorithm: a feasibility study [J]. Phys Med Biol, 2016, 61: 6553- 6569.
[18] Zhang L, Fried DV, Fave XJ, et al. IBEX: an open infrastructure software platform to facilitate collaborative work in radiomics [J]. Med Phys, 2015, 42: 1341- 1353.
[19] Parmar C, Grossmann P, Bussink J, et al. Machine learning methods for quantitative radiomic biomarkers [J]. Sci Rep, 2015, 5: 13087.
[20] Liehr T, Acquarola N, Pyle K, et al. Next generation phenotyping in emanuel and pallister-killian syndrome using computer-aided facial dysmorphology analysis of 2D photos [J]. Clin Genet, 2018,93:378- 381.
[21] Katsanis SH,Katsanis N. Molecular genetic testing and the future of clinical genomics [J]. Nat Rev Genet, 2013, 14: 415- 426.
[22] Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for mendelian disease gene discovery [J]. Nat Rev Genet, 2011, 12: 745- 755.
[23] Fernandez-Marmiesse A, Gouveia S,Couce ML. NGS technologies as a turning point in rare disease research, diagno-sis, and treatment [J]. Curr Med Chem, 2018,25:404- 432.
[25] Visscher PM, Brown MA, McCarthy MI, et al. Five years of GWAS discovery [J]. Am J Hum Genet, 2012, 90: 7- 24.
[26] Denny JC, Bastarache L, Roden DM. Phenome-wide association studies as a tool to advance precision medicine [J]. Annu Rev Genomics Hum Genet, 2016, 17: 353- 373.
[27] Visscher PM, Wray NR, Zhang Q, et al. 10 Years of GWAS discovery: biology, function, and translation [J]. Am J Hum Genet, 2017, 101: 5- 22.
[28] Denny JC, Ritchie MD, Basford MA, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations [J]. Bioinformatics, 2010, 26: 1205- 1210.
[29] Bush WS, Oetjens MT,Crawford DC.Unravelling the human genome-phenome relationship using phenome-wide association studies [J]. Nat Rev Genet, 2016, 17: 129- 145.
[30] McKusick VA. Mendelian Inheritance in Man and its online version, OMIM [J]. Am J Hum Genet, 2007, 80: 588- 604.
[31] Pavan S, Rommel K, Mateo Marquina ME, et al. Clinical practice guidelines for rare diseases: the orphanet database [J]. PLoS One, 2017, 12: e0170365.
[32] Swaminathan GJ, Bragin E, Chatzimichali EA, et al. DECIPHER: web-based, community resource for clinical interpretation of rare variants in developmental disorders [J]. Hum Mol Genet, 2012, 21: R37- R44.
[33] Zhang X, Minikel EV, O’Donnell-Luria AH, et al. ClinVar data parsing [J]. Wellcome Open Res, 2017, 2: 33.
[34] Stenson PD, Ball EV, Mort M, et al. The human gene mutation database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution [J]. Curr Protoc Bioinformatics, 2012, Chapter 1: Unit1.13.
[35] Stelzer G, Rosen N, Plaschkes I, et al. The gene cards suite: from gene data mining to disease genome sequence analyses [J]. Curr Protoc Bioinformatics, 2016, 54:1.30.1- 1.30.33.
[36] Rappaport N, Twik M, Plaschkes I, et al. MalaCards: an amalgamated human disease compendium with diverse clinical and genetic annotation and structured search [J]. Nucleic Acids Res, 2017, 45: D877-D887.
[37] Pepin MG, Murray ML, Bailey S, et al. The challenge of comprehensive and consistent sequence variant interpretation between clinical laboratories [J]. Genet Med, 2016, 18: 20- 24.
[38] Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [J]. Genet Med, 2015, 17: 405- 424.
[39] Van Dijk EL, Auger H, Jaszczyszyn Y, et al. Ten years of next-generation sequencing technology [J]. Trends Genet, 2014, 30: 418- 426.
[40] Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a mendelian disorder [J]. Nat Genet, 2010, 42: 30- 35.
[41] Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome [J]. Nat Genet, 2010, 42: 790- 793.
[42] Hoischen A, van Bon BW, Gilissen C, et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome [J]. Nat Genet, 2010, 42: 483- 485.
[43] Gilissen C, Arts HH, Hoischen A, et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome [J]. Am J Hum Genet, 2010, 87: 418- 423.
[44] Beaulieu CL, Majewski J, Schwartzentruber, et al. FORGE Canada Consortium: outcomes of a 2-year national rare-disease gene-discovery project [J]. Am J Hum Genet, 2014, 94: 809- 817.
[45] Welter D, MacArthur J, Morales J, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations [J]. Nucleic Acids Res, 2014, 42: D1001-D1006.
[46] Duan L, Wei L, Tian Y, et al. Novel susceptibility loci for moyamoya disease revealed by a genome-wide association study [J]. Stroke, 2018, 49: 11- 18.
[47] Sardana D, Zhu C, Zhang M, et al.Drugrepositioningforor-phandiseases[J]. Brief Bioinform, 2011,12: 346- 356.
[48] Stockklausner C, Lampert A, Hoffmann GF, et al. Novel treatments for rare cancers: the U.S. orphan drug act is deli-vering-A cross-sectional analysis [J]. Oncologist, 2016, 21: 487- 493.
[49] Zhao M,Wei DQ. Rare diseases: drug discovery and informatics resource [J]. Interdiscip Sci, 2018,10:195- 204.
[50] Kodra Y,Posada de la Paz M, Coi A,et al. Data quality in rare diseases registries [J]. Adv Exp Med Biol, 2017, 1031: 149- 164.
