玉米株型相关性状的全基因组关联分析
2018-03-24刘坤张雪海孙高阳闫鹏帅郭海平陈思远薛亚东郭战勇谢惠玲汤继华李卫华
刘坤,张雪海,孙高阳,闫鹏帅,郭海平,陈思远,薛亚东,郭战勇,谢惠玲,汤继华,李卫华
玉米株型相关性状的全基因组关联分析
刘坤1,张雪海1,孙高阳1,闫鹏帅1,郭海平1,陈思远2,薛亚东1,郭战勇1,谢惠玲1,汤继华1,李卫华1
(1河南农业大学农学院/省部共建小麦玉米作物学国家重点实验室,郑州 450002;2郑州市第一中学,郑州 450002)
【目的】玉米株型性状与植株产量、光合效率、抗倒性等密切相关,是理想株型设计育种的基础,通过对玉米多个株型相关性状进行全基因组关联分析,构建玉米理想株型,为抗倒伏、耐密性的玉米新品种选育提供理论基础。【方法】以284份温带、亚热带和热带材料组成的关联群体为研究对象,在郑州与浚县2个环境下对玉米总叶片数(LN)、穗上叶片数(LNAN)、穗上叶片数与总叶片数比值(LNAN/LN)、株高(PH)、穗位高(EH)、穗位高与株高的比值(EH/PH)等6个玉米株型相关性状进行调查,借助覆盖玉米全基因组约56万个SNP标记,进行全基因组关联分析,以期为玉米新品种的选育提供理论依据。【结果】2个环境下,6个性状均表现为正态分布;大多数性状之间均存在高度正相关或负相关;方差分析表明这6个株型相关性状的基因型与环境以及基因型×环境的互作均达到显著水平。在选择最优关联分析模型时,发现Q模型假阳性较高,Q+K模型对假阳性的控制过于严格,而K模型的表现最好;在2个环境下,全基因组关联分析(K模型)共检测到56个显著SNP-性状关联(≤3.99E-6),这些SNPs共涉及5个性状的17个位点,每个位点解释的表型变异从7.97%—10.56%不等。同时发现有4个位点能够在2个环境中同时被检测到,表明这4个位点受环境影响较小,在不同环境下可以稳定存在。通过对显著关联的SNP上下游各50 kb范围内候选基因进行搜索,共发现80个候选基因,其中42个具有功能注释。例如,与株高和穗位高显著相关的基因GRMZM2G161293编码一个具有乙酰葡糖转移酶活性的蛋白,该酶催化UDP-N-乙酰氨基葡萄糖生成糖过程中的N-乙酰葡糖氨基残基的转移,可能通过影响玉米籽粒可溶性糖含量进而影响产量,推测其为最可能的候选基因。【结论】K模型对假阳性的控制效果最好,基于K模型的GWAS结果,一共检测到17个株型性状相关的位点。
玉米(L.);全基因组关联分析(GWAS);株型性状;理想株型
0 引言
【研究意义】玉米的理想株型[1]具有较高的光能利用效率,具备制造更多干物质的生理潜力。实现生育期内源、流和库的协调发展,合理分配光合产物,可使植株间的竞争达到最小状态,从而提高产量。株型相关性状如株高、穗位高、叶片数等与植株产量、光合效率、抗倒性等密切相关,是理想株型设计育种的基础。因此,对其遗传基础进行研究,不但可以了解玉米理想株型建成的遗传机制,而且还可帮助在育种实践中有效降低植株高度与穗位高度、合理调控叶片数目及分布,从而提高作物产量。【前人研究进展】玉米株型性状是典型的数量性状,由多基因控制并受多种因素尤其是环境的影响十分显著。因此,科研工作者常利用数量性状位点(quantitative trait locus,QTL)定位的方法分析玉米株型性状的遗传机制,解析基因的遗传效应、基因之间的互作及基因与环境之间的互作关系。近年来,随着测序技术和统计方法的发展,研究者借助高密度标记分别对株高、穗位高、叶夹角、叶长、叶宽、节间数等玉米株型性状的遗传规律进行解析。玉米株高与穗位高是株型的重要构成因子,并且是玉米数量遗传学研究的模式性状,前人分别利用不同的分离群体(如重组自交系群体)和关联群体等[2-8],对玉米株高和穗位高的遗传机制进行了大量研究,检测到多个与株高和穗位高相关的QTL,分布在玉米的10条染色体上[9-10]。其中位于玉米第1染色体的所在的玉米株高效应位点,可以解释20%的表型变异。Teng等[11]克隆的株高基因,被认为是调控玉米株高的主效QTL即的候选基因。Yang等[12]对玉米株高进行了关联分析,在混合线性模型下仅找到一个位于第3染色体的SNP(chr.3:162709488),该SNP可解释9.61%的表型变异;Pan等[13]利用3种不同的定位方法(SLM、JLM和GWAS)对10个重组自交系群体组成的ROAM群体的株型性状进行定位,在SLM、JLM和GWAS下分别鉴定到83、86和38个株高QTL,并精细定位了一个位于第3染色体的株高主效QTL到600 kb(165.73—166.37 Mb)。国内外许多学者对玉米叶片数与产量之间的关系也进行了大量研究[14-16],发现虽然多叶品种可以增加穗上部分的叶片面积,增加了受光面积指数,使得植株生物量增高,但是多叶品种并没有增加籽粒产量。而玉米品种最终产量的提高是在种植密度不变的条件下增加了玉米的叶面积指数,进而增加受光面积,加速灌浆速率[17-20],其中穗上叶片数对单产有较大的正向直接效应[21]。【本研究切入点】尽管前人利用不同的分离群体定位并克隆了一些与株高、穗位高、叶片数等株型性状相关的QTL或基因,但由于这些性状属于复杂的数量性状,人们对其遗传机理还知之甚少。【拟解决的关键问题】本研究以284份遗传多样性丰富的温带、亚热带和热带材料组成的关联群体为研究对象,借助覆盖玉米全基因组的约56万个SNP标记,对玉米多个株型相关性状进行全基因组关联分析,旨在为玉米理想株型的构建,为抗倒伏、耐密植的玉米新品种选育提供理论基础。
1 材料与方法
1.1 试验材料
所用材料为华中农业大学严建兵教授提供,是从关联群体(n=513)中选择的在郑州及鹤壁适应性较好的284份玉米自交系构成,主要来源为热带(104份)、亚热带(42份)及温带材料(138份)。
1.2 田间试验设计
关联群体于2012年分别种植在郑州河南农业大学试验站(郑州试验站)与鹤壁浚县的鹤壁农科院试验站(浚县试验站)。田间设计采用完全随机区组设计,3次重复,每个材料单行区种植,行长4 m,行距0.67 m,株距0.25 m,密度63 000株/hm2,田间管理采用常规管理。为了叶片数目调查的准确性,在小喇叭口和大喇叭口时期分别对玉米第5和第10片叶进行标记,并在玉米灌浆末期,每一行挑选10株长势均匀的植株对4个株型相关性状株高(plant height,PH)、穗位高(ear height,EH)、总叶片数(leaf number,LN)和穗上叶片数(leaf number above ear,LNAN)进行调查。
1.3 数据处理与分析
1.3.1 一般统计分析 采用Excel、SPSS19.0统计软件对2个环境下关联群体的株高(PH)、穗位高(EH)、总叶片数(LN)、穗上叶片数(LNAN)以及由此衍生的穗位高/株高(EH/PH)和穗上叶片数/总叶片数(LNAN/LN)等6个性状进行统计分析,并分别计算各性状的平均值、标准差、标准误、频率分布等,并进行相关性分析。
同时根据R语言的lme4包[22]的混合线性模型进行最佳线性无偏预测(best linear unbiased prediction,Blup),评估2个环境下每一个材料每一个性状的育种值,最终Blup值也用于后续的全基因组关联分析。
1.3.2 全基因组关联分析 所使用的基因型为华中农业大学严建兵教授提供的覆盖玉米全基因组的最小等位基因频率大于0.05的558 629个SNPs组成[12],基因型数据可在网站http://www.Maizego.org/Resources.html下载。考虑到很多标记间可能存在高度连锁不平衡,因此,采用GEC软件[23]计算了有效标记数(effective number,Ne),并根据软件给出的建议值(1/Ne)作为确定各性状-SNP间是否显著关联的依据。
在全基因组关联分析(GWAS)中,由于不同性状对GWAS不同模型控制假阳性和假阴性的敏感程度不同,因此统计功效是首先要考虑的因素。为了选择最优的GWAS模型,使用了3种常用的模型,即只控制群体结构的Q模型(Q)、只控制亲缘关系的K模型(K)及同时控制群体结构和亲缘关系的Q+K模型(Q+K),对每一个性状进行了GWAS分析。通过对3种模型的QQ Plot的比较来选择最优模型并对最优模型的GWAS结果进行进一步解析,以上GWAS分析均在TASSEL3.0(http://www.maizegenetics.net/)软件中完成。先前研究表明,该群体的连锁不平衡(linkage disequilibrium,LD)衰减距离为50 kb[24-25],利用558 629个SNP标记对所用材料的LD进行了重新评价,发现平均LD为50—100 kb(2=0.1),简便起见,选择50 kb作为LD衰减距离。因此定义峰值SNP上下游各50 kb的区间范围(即共100 kb)为一个位点;同时定义不同位点间,若存在重叠部分,则这些位点构成一个非重复的QTL。另外,基于B73参考基因组(B73 RefGen_v2),从玉米数据库MaizeGDB(http://www.maizegdb.org)下载玉米基因组的基因列表搜索每一个位点内的所有可能的候选基因。根据InterProScan数据库(http: //www. ebi.ac.uk/ interpro/scan.html)并结合美国国立生物技术信息中心数据库(https://www.ncbi.nlm.nih.gov/)对每一个候选基因进行功能注释。最后根据每一个位点内所有基因的功能注释及其在B73的各组织的表达情况,选择一个最可能的候选基因。
2 结果
2.1 株型相关农艺性状表型数据分析
对此关联群体在不同环境下的6个株型相关性状LN、LNAN、LNAN/LN、PH、EH、EH/PH的表型进行统计分析(表1),得知玉米株型相关性状存在广泛变异。同时,也发现浚县试验站的株高、穗位高、总叶片数及穗上叶片数均高于郑州试验站(图1),主要是由于浚县位于更高纬度,日照长度要高于郑州,导致营养生产期延长所致,同时发现2个环境中株型相关性状的峰度和偏度的绝对值均小于1(表1),暗示这些性状均遵从正态分布,表明这些性状均为典型的数量性状,由微效多基因控制。
2.2 株型相关性状的相关性分析和方差分析
通过对2个环境的6个玉米株型性状进行相关性分析,发现除了浚县试验站穗位高与穗上叶片数及2个环境下株高与穗位高/株高的比值不存在显著相关之外。其余各性状之间均表现出显著正相关或显著负相关(表2)。2个环境中的穗上叶片数与总叶片数的比值和穗上叶片数之间、以及穗位高与株高比值和穗位高之间、株高和穗位高之间的相关系数较大,并处于极显著水平。说明各性状的发育存在着相互协同促进的作用。
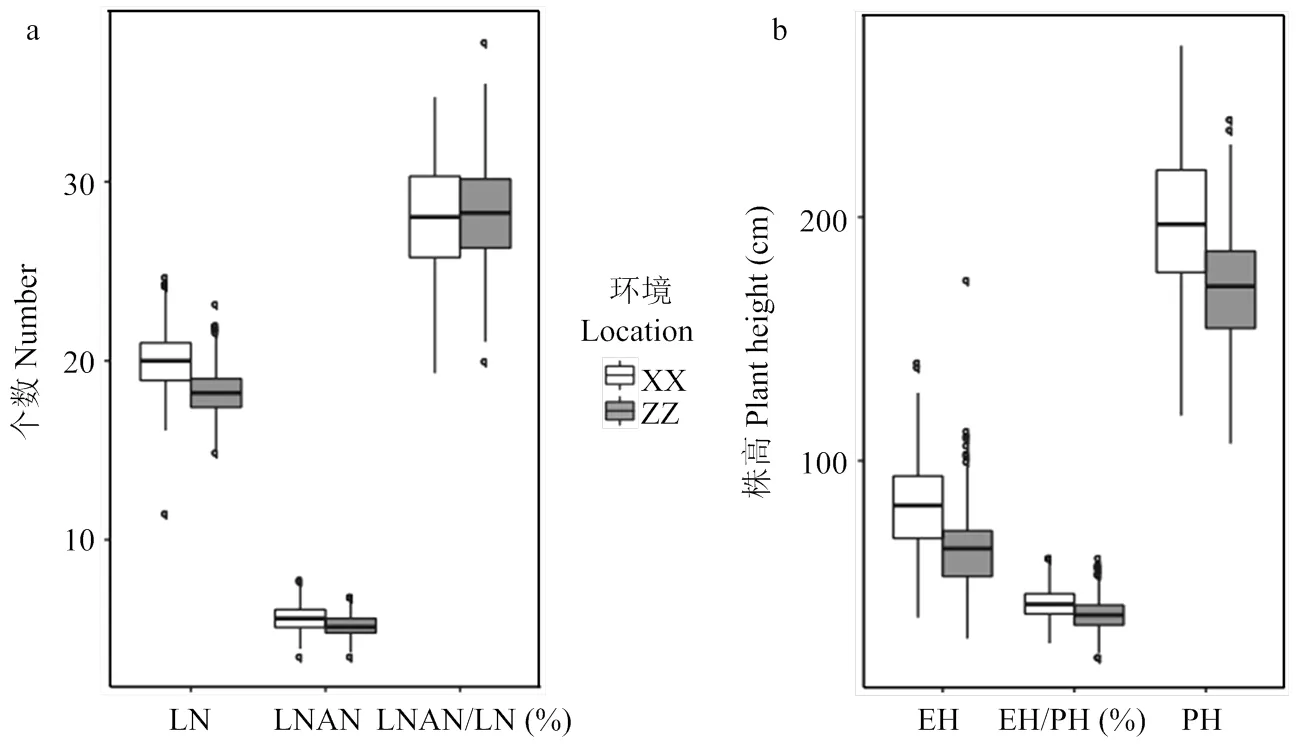
a:叶片数相关性状;b:株高相关性状;XX表示浚县试验站;ZZ表示郑州试验站;LN:总叶片数;LNAN:穗上叶片数;LNAN/LN表示穗上叶片数与总叶片数比值;EH:穗位高;PH:株高;EH/PH:穗位高与株高的比值。下同
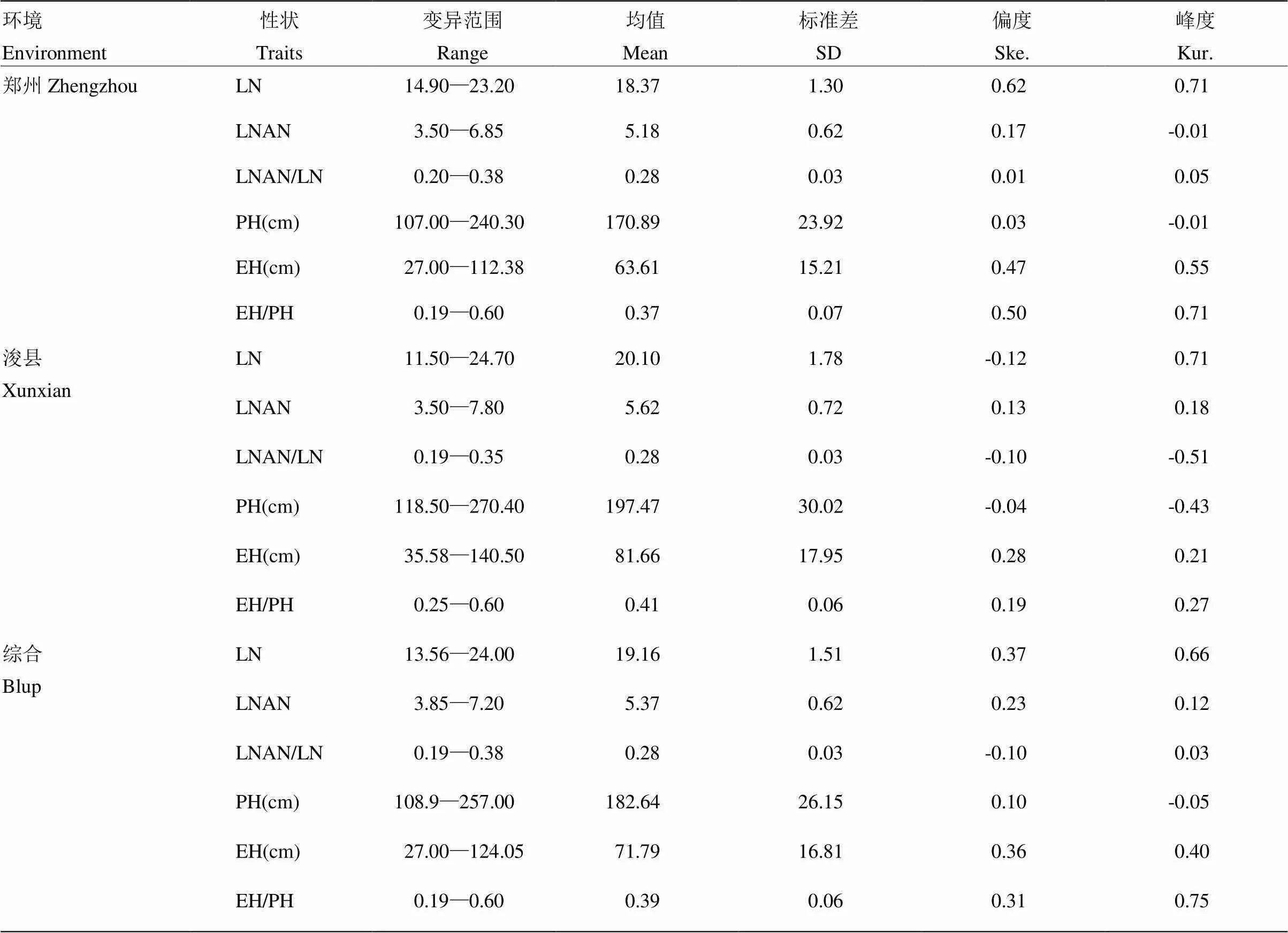
表1 玉米不同环境下株型相关性状描述统计分析

表2 关联群体株型性状的相关性分析
**:在0.01水平上显著相关;*:在0.05水平上显著相关。下同。对角线右上角表示浚县点,对角线左下角表示郑州点
* *: Correlation is significant at the 0.01 level; *: Correlation is significant at the 0.05 level. The same as below. Correlation coefficients in Xunxian are above the diagonal, in Zhengzhou are below the diagonal
方差分析结果表明,关联群体所有6个株型性状的基因型效应都达到极显著水平,说明关联群体各材料间的株型性状存在显著的遗传变异(表3)。同时,环境间、基因型与环境互作的差异也达到极显著水平,说明玉米株型不同性状受环境影响较大,同时也存在着基因型与环境之间的互作关系。
2.3 株型相关性状全基因组关联分析
2.3.1 阈值确定 通过GEC软件[23]计算了558 629个SNPs的有效标记数,结果表明,有效标记数Ne为250 345,软件给出的建议值为3.99E-6(1/Ne),因此,该值将作为后续GWAS分析的标记-性状间显著关联的阈值。
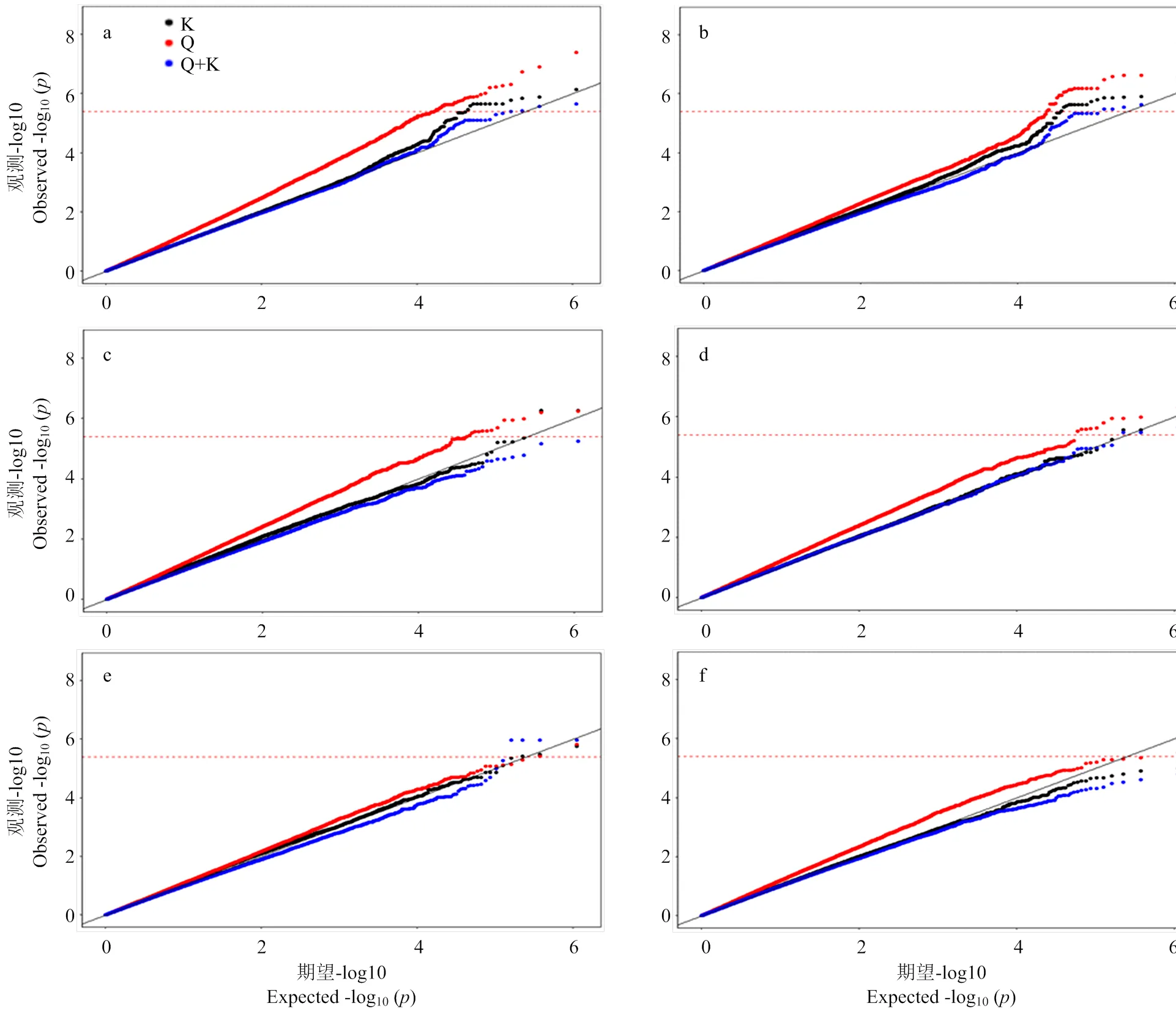
2.3.2 模型选择 以2个环境下6个性状的Blup值(作为一个新的环境)为例,用3种模型(Q、K和Q+K)分别进行了GWAS分析,通过不同性状3种模型的比较,从QQ图中不难看出(图2),对这6个性状来说,Q模型(图中红色点)对假阳性的控制效果最差,而K(图中黑色点)和Q+K模型(图中蓝色点)对假阳性的控制较好,但是Q+K模型控制的过于严格,综合考虑,在后续分析中,对这6个性状,均选择K模型的结果进行进一步阐述。
2.3.3 关联分析结果解析 利用覆盖全基因组的558 629个SNP标记对关联群体在2个环境下的6个株型性状进行关联分析,基于K模型,在≤3.99E-6下,6个株型性状一共检测到56个SNPs-性状关联。2个环境联合分析条件下(Blup),6个株型相关性状的曼哈顿图见图3。
用55万SNPs标记对该群体LD的评估表明,该群体的衰减距离为50 kb[24],基于此衰减距离,56个SNPs-性状关联涉及到的SNP一共分布于17个位点内,该结果与数量性状位点的本质相一致,即由微效多基因控制;其中在郑州试验站、浚县实验站及Blup下分别检测到8个、5个和9个位点。具体来说(表4和图4),郑州环境下一共检测到10个SNP-性状关联,涉及到8个位点,分别是位于第5和第8染色体与株高有关的2个位点,分别解释8.73%和9.54%的株高表型变异;位于玉米第3、5、6、8染色体与穗位高有关的4个位点,每个位点解释的表型变异在8.19%—10.10%不等,平均为8.83%;及位于第4和第8染色体与穗位高与株高比值相关的2个位点,分别解释8.83%和10.42%的表型变异。并没有检测到与叶片数相关性状的显著位点。而在浚县环境下,一共检测到5个显著位点,分别为与株高有关的位于第1和第7染色体的2个位点,分别可解释9.69%和9.66%的表型变异;及与穗位高,穗位高与株高比值及总叶片数有关的分别位于第8、7、10染色体的3个位点,这3个位点分别能解释10.31%、9.34%和10.46%表型变异。2个环境联合分析(Blup)下,在第9染色体共检测到与穗位高相关的8个显著位点(涉及到16个显著的SNP),每个位点平均可解释8.76%的表型变异;同时发现位于第5、8、9染色体的3个与株高有关的位点,分别解释8.12%、9.20%和8.58%的表型变异,其中位于第9染色体的位点里有11个显著的SNPs,并且该位点与穗位高的位点为同一个位点。还发现1个位于第8染色体的与穗位高与株高比值的位点,可解释10.12%的表型变异;2个与总叶片数有关的位点,位于第7和第10染色体,分别解释8.12%和8.22%的表型变异;另外还发现2个分别位于第3和第5与穗上叶片数有关的位点,分别可解释8.06%和8.88%的表型变异。
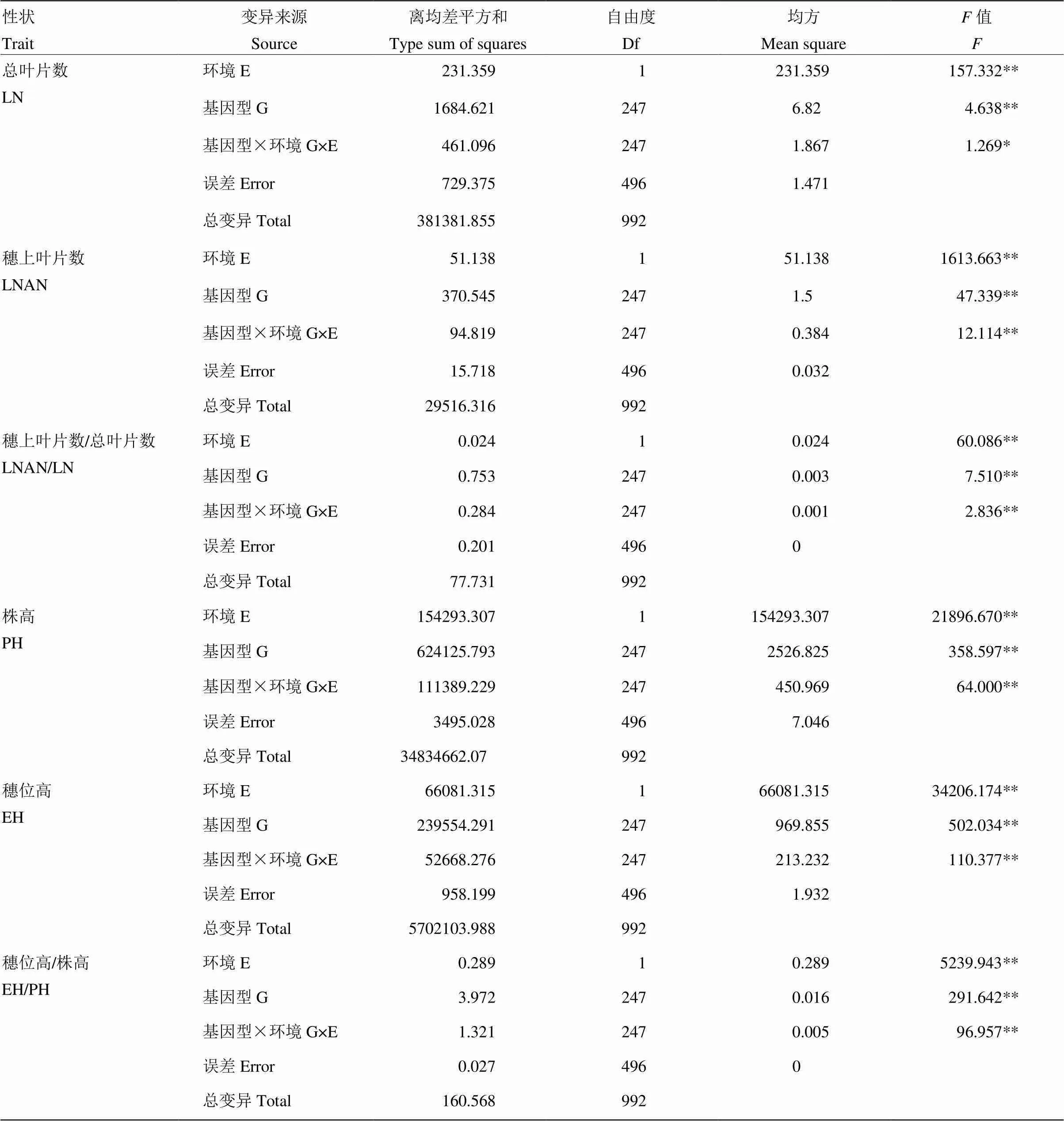
表3 玉米不同环境下株型相关性状的方差分析
E:环境;G:基因型;G×E:基因与环境互作
E: environment; G: genotype; G×E: genotype-by-environment interaction
同时发现有4个位点,分别为表4中的第7、13、14和17位点可同时在2个以上环境中被检测到,表明这些位点受环境影响较小,可在不同环境下稳定遗传,这些信息为将来继续深入分析这些位点提供了有价值的信息。值得注意的是,在2个环境及Blup下均没检测到与穗上叶片数与总叶片数比值相关的位点(图3、图4和表4),表明这一性状受环境影响较大或在该群体中不存在控制这一性状的主效位点。
a:株高_2012;b:穗位高_2012;c:穗位高/株高_2012;d:穗上叶片数_2012;e:总叶片数_2012;f:穗上叶片数/总叶片数_2012。下同
2.4 株型相关候选基因确定
基于该群体的LD衰减距离,在峰值SNP上下游各50 kb范围内搜索候选基因。6个株型性状的17个显著关联位点上下游各50 kb范围内一共搜到80个候选基因,其中有功能注释的基因有42个。根据每一个位点内所有候选基因的功能注释及其在B73的各组织的表达情况,选择一个最可能的候选基因及其注释列于表4。
3 讨论
由于不同性状对GWAS不同模型在控制Ⅰ型错误(假阳性)的敏感性不同,即存在性状依赖性,因此,在进行GWAS分析前首要考虑的是GWAS的统计功效,即最大可能的降低假阳性。先前研究表明,对于玉米的开花期、穗位高和穗粗性状来说,K模型和Q+K模型均能很好的控制假阳性,但是Q+K模型比K模型更好[26]。本研究发现Q+K模型对假阳性的控制较严格(图2),若采用Q+K模型则会产生更多的假阴性结果,而K模型对假阳性的控制效果最好,因此,最终选择表现较好的K模型对GWAS结果进行进一步解析。
玉米的农艺及产量相关性状的研究近年来报道较多,陈荣江等[27]通过研究玉米的穗长、穗行数、行粒数、百粒重、株高、穗位高、穗重及穗粗在内的8个性状与产量之间的关系,发现这8个性状与产量存在正相关;汤继华等[2]以豫玉22构建的260多个F2:3家系为材料,研究了不同环境条件下的穗位高、雄穗分枝数、茎粗与产量之间的关系,发现与产量之间也存在显著正相关。本研究以284份遗传性丰富的温带、热带、亚热带材料为研究对象,在郑州和浚县2个环境下对玉米株高、穗位高、总叶片数和穗上叶片数以及株高/穗位高、总叶片数/穗上叶片数等6个株型性状进行分析,发现在2个环境下的不同性状之间均存在显著正相关或者负相关,说明各性状的发育存在着相互协同促进作用。通过对郑州和浚县这6个株型性状的全基因组关联分析,在K模型下,一共检测到56个SNP-性状关联(株高、穗位高、穗位高与株高比值、总叶片数、穗上叶片数),这些SNP涉及到17个位点。而穗上叶片数与总叶片数比值这一性状没有检测到显著的SNPs,表明该性状受环境影响较大或者在该关联群体中不存在控制这一性状的主效位点。在这17个位点里,同时发现有4个位点能够在2个环境中被检测到,表明这4个位点受环境影响较小,在不同环境下可以稳定存在。同时也发现,有的性状和多个位点显著关联,表明这些性状受微效多基因控制,符合数量性状的本质[28]。
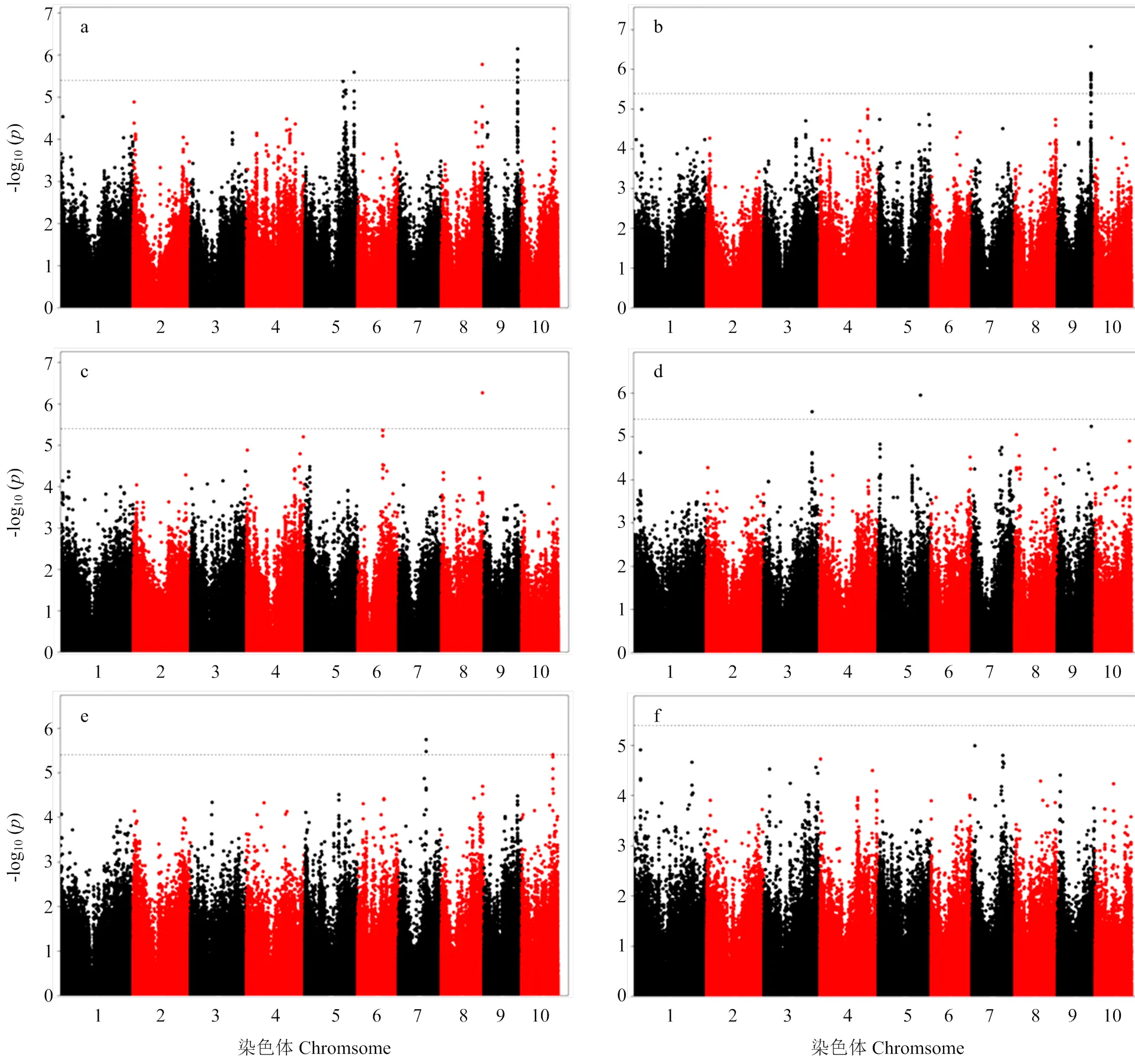
图3 浚县与郑州2个环境结合分析(Blup)下株型相关性状的曼哈顿图
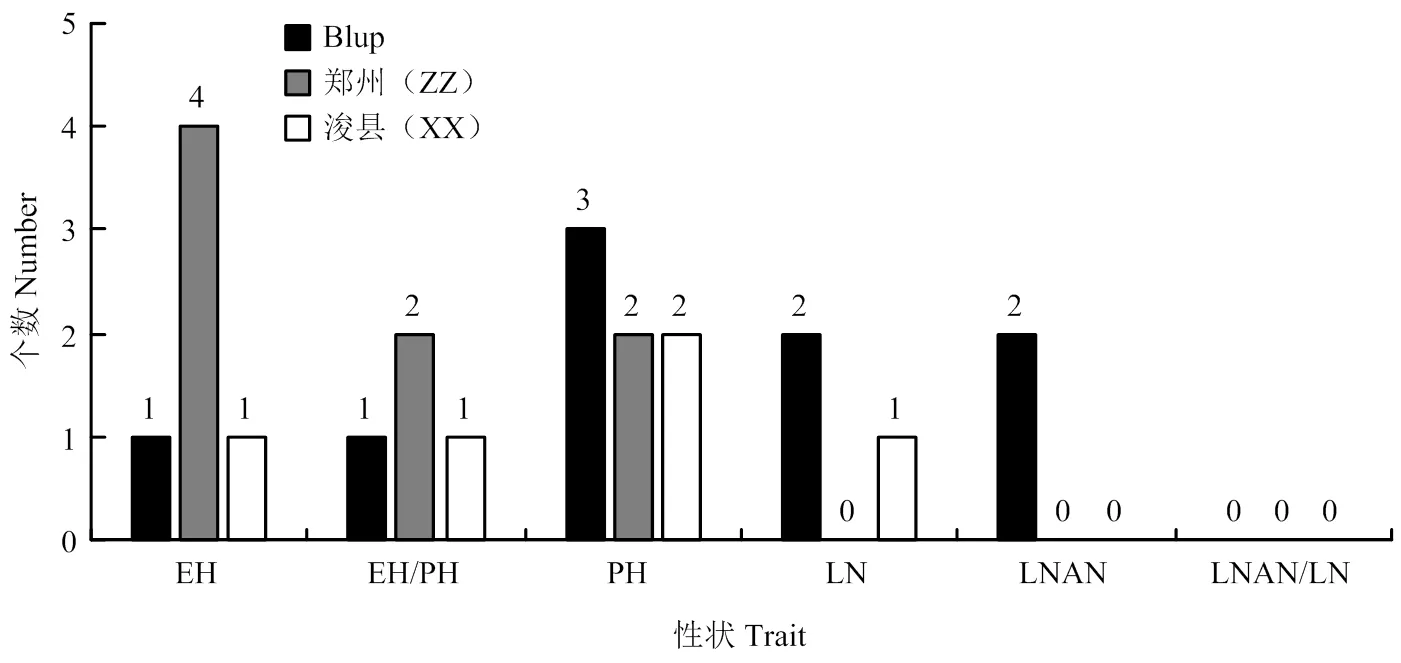
图4 各个环境下株型相关性状显著位点的分布
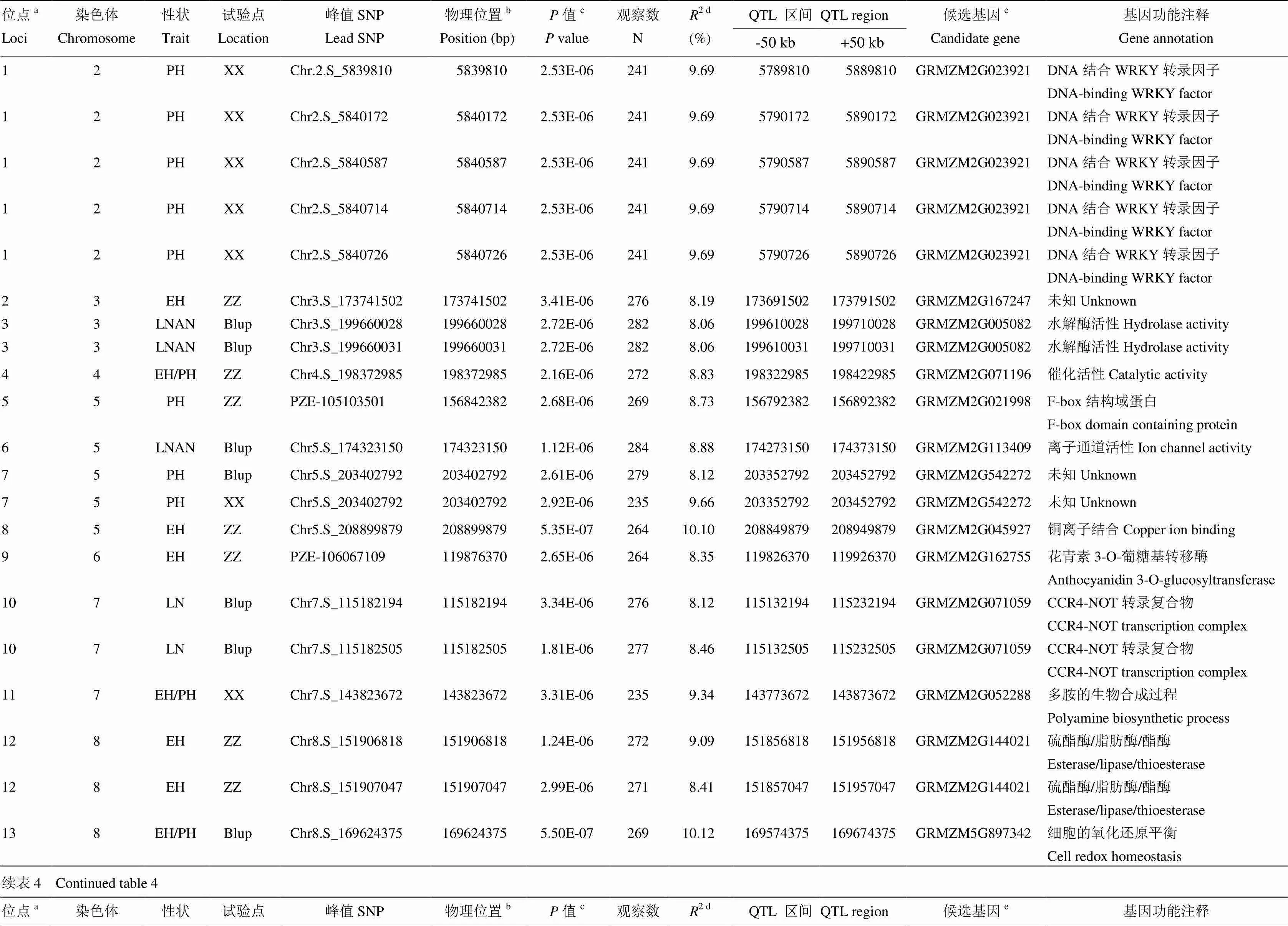
表4 全基因组关联分析鉴定到的显著位点及有关信息以及每个位点里的最可能的候选基因及其功能注释
a所有有重叠区间的QTL被归为一个非重复的QTL或位点,位点的ID根据染色体及物理位置升序排序命名;b每个SNP的物理位置是基于B73参考基因组v2版本得到;c相应性状的值是通过K模型计算得到;d相应位点解释的表型变异;e位点内一个可能的生物学候选基因或离峰值SNP最近的候选基因。ZZ表示郑州,XX表示浚县,Blup表示2个环境的综合
aAll QTLs with overlapping QTL regions were categorized as a non-redundant QTL or loci, ID of loci was numbered according to chromosome and position by a ascending order method;bPhysical position of each SNP based on B73 RefGen_v2.cP value of the corresponding trait calculated by K model.dThe phenotypic variance explained by the corresponding locus.eA plausible biological candidate gene in the locus or the nearest annotated gene to the lead SNP. ZZ: Zhengzhou; XX: Xunxian
基于B73参考基因组及每一个显著位点的LD衰减距离,在这17个位点里一共发现80个候选基因,其中42个基因有功能注释,其余38个基因的功能未知;42个已知功能的基因涉及到转导蛋白、还原蛋白、金属离子转运蛋白、脂类蛋白、纤维素合酶等,功能涉及能量代谢、物质运输、生物调控、信号传导等生化代谢途径。
17个位点里,发现多个位点里存在多个显著的SNP,比如位点16里含有17个显著SNP,表明这些SNP间可能存在很强的连锁不平衡关系,这些成簇SNP所在区域,是控制株型性状的热点区域,有必要对其加以深入挖掘,为培育理想株型品种的选育提供重要研究价值。郑克志等[25, 29]以玉米自交系T319与9406为亲本构建的242个重组自交系(F8)对玉米株高和穗位高进行QTL分析,结果发现6个株高相关的QTL,其中有2个QTL同时控制穗位高。本研究也发现了4个位点(位点7、13、14和17)可同时在2个以上环境中被检测到(表4),表明这些位点受环境影响较小,可在不同环境下稳定遗传,同时也发现有些位点同时控制多个性状,比如位点16同时控制株高和穗位高,这些信息为将来继续深入利用和克隆这些位点提供了有价值的信息。
本研究通过玉米株型性状显著关联的SNP所在位点对可能的候选基因进行预测。6个性状,有5个性状均能检测到显著位点,随后对每一个性状的显著位点的候选基因及其功能进行了预测(表4),值得注意的是,在郑州和浚县2个环境的综合条件下,位于第9染色体的位点16(chr.9:139516295—chr.9:139517032)同时与株高和穗位高显著关联,并且该位点在先前的两个研究中也被报道与株高相关(假定2个研究定位的显著SNP物理位置小于5 Mb被认为同一个位点)。比如,Yang等[12]通过A—D test共检测到株高相关的107个显著SNP,其中第9染色体的4个SNP(chr9..S_140356597、chr.9.S_140435314、chr.9.S_142397931和chr.9.S_142495942)与该位点共定位;Samayoa等[30]通过关联分析检测到18个株高显著SNP,其中第9染色体的一个SNP, S9_142752809,与该位点共定位;该位点(位点16)内一共存在4个候选基因(GRMZM5G872568、GRMZM2G063875、GRMZM2G161293和GRMZM2G366532),除GRMZM2G161293外,其余基因的功能未知。而GRMZM2G161293编码一个BGGP Beta-1-3-半乳糖-O-糖基-糖蛋白,具有乙酰葡糖转移酶活性,该酶催化UDP-N-乙酰氨基葡萄糖生成糖过程中的N-乙酰葡糖氨基残基的转移,猜测可能通过影响玉米籽粒可溶性糖含量进而影响产量,推测其为最可能的候选基因;另外发现在2个环境下能同时检测到的4个位点(位点7、13、14和17),其中基因GRMZM2G005082、GRMZM5G897342、GRMZM2G440125和GRMZM2G074689分别位于位点7、13、14和17中,这4个基因分别编码水解酶,常温蛋白,锌离子结合蛋白及钙离子结合蛋白;这些同时控制多个性状的SNP或位点及其在2个环境中同时被检测到的SNP或位点可能与株型相关性状的候选基因紧密连锁,可以作为下一步研究的重点区域。
4 结论
本研究借助覆盖玉米全基因组的约56万个SNP标记,利用3种模型(Q、K和Q+K)对玉米总叶片数(LN)、穗上叶片数(LNAN)、穗上叶片数与总叶片数比值(LNAN/LN)、株高(PH)、穗位高(EH)、穗位高与株高的比值(EH/PH)等6个玉米株型相关性状进行全基因组关联分析,发现K模型对假阳性的控制效果最好。基于K模型的GWAS结果,一共检测到56个SNP-性状关联,涉及17个位点,对显著关联的SNP上下游各50 kb范围内候选基因进行搜索,共发现80个候选基因,其中42个具有功能注释。
致谢:华中农业大学作物遗传改良国家重点实验室严建兵教授为本试验提供关联群体材料及基因型,在此表示感谢。
[1] Donald M. Breeding of crop ideotypes., 1968, 17(3): 385-403.
[2] 汤继华, 谢惠玲, 黄绍敏, 胡彦民, 刘宗华, 季洪强, 寇志安. 缺氮条件下玉米自交系叶绿素含量与光合效率的变化. 华北农学报, 2005, 20(5): 10-12.
TANG J H, XIE H L, HUANG S M, HU Y M, LIU Z H, JI H Q, KOU Z A. The changes of the content for chlorophyll and photosynthetic productivity in maize inbred lines under the low-nitrogen stress., 2005, 20(5): 10-12. (in Chinese)
[3] 何坤辉, 常立国, 崔婷婷, 渠建洲, 郭东伟, 徐淑兔, 张兴华, 张仁和, 薛吉全, 刘建超. 多环境下玉米株高和穗位高的QTL定位. 中国农业科学, 2016, 49(8): 1443-1452.
HE K H, CHANG L G, CUI T T, QU J Z, GUO D W, XU S T, ZHANG X H, ZHANG R H, XUE J Q, LIU J C. Mapping QTL for plant height and ear height in maize under multi-environments., 2016, 49(8): 1443-1452. (in Chinese)
[4] 殷鹏程. 玉米株高和穗位高的QTL定位[D]. 武汉: 华中农业大学, 2015.
YIN P C.QTL mapping of plant height and ear height in maize[D]. Wuhan: Huazhong Agricultural University, 2015. (in Chinese)
[5] 杨俊品, 荣廷昭, 向道权, 唐海涛, 黄烈建, 戴景瑞. 玉米数量性状基因定位. 作物学报, 2005, 31(2): 188-196.
YANG J P, RONG T Z, XIANG D Q, TANG H T, HUANG L J, DAI J R. QTL mapping of quantitative traits in maize., 2005, 31(2): 188-196. (in Chinese)
[6] 曹永国, 王国英, 王守才, 魏艳玲, 卢江, 谢友菊, 戴景瑞. 玉米RFLP遗传图谱的构建及矮生基因定位. 科学通报, 1999, 44(20): 2178-2182.
CAO Y G, WANG G Y, WANG S C, WEI Y L, LU J, XIE Y J, DAI J R. Construction and localization of dwarf gene RFLP genetic map of maize., 1999, 44(20): 2178-2182. (in Chinese)
[7] 兰进好, 李新海, 高树仁, 张宝石, 张世煌. 不同生态环境下玉米产量性状QTL分析. 作物学报, 2005, 31(10): 1253-1259.
LAN J H, LI X H, GAO S R, ZHANG B S, ZHANG S H. QTL analysis of yield components in maize under different environments., 2005, 31(10): 1253-1259. (in Chinese)
[8] 许诚, 王彬, 毛克举, 胡彦民, 谢惠玲, 汤继华. 利用单片段代换系群体定位玉米株型性状QTL. 玉米科学, 2014, 22(2): 28-34.
XU C, WANG B, MAO K J, HU Y M, XIE H L, TANG J H. QTL mapping for plant-type related traits using single segment substitution lines in maize., 2014, 22(2):28-34. (in Chinese)
[9] 张志明, 赵茂俊, 荣廷昭, 潘光堂. 玉米SSR连锁图谱构建与株高及穗位高QTL定位. 作物学报, 2007, 33(2): 341-344.
ZHANG Z M, ZHAO M J, RONG T Z, PAN G T. SSR linkage map construction and qtl identification for plant height and ear height in maize (L)., 2007, 33(2): 341-344. (in Chinese)
[10] Xing A, Gao Y, Ye L F,CAI L CHING A, LLACA V, JOHNSON B, LIU L, YANG X H, KANG D M, YAN J B, LI J S. A rare SNP mutation in brachytic2 moderately reduces plant height and increases yield potential in maize., 2015, 66(13): 3791-3802.
[11] Teng F, Zhai L, Liu R, BAI W, WANG L, HUO D, TAO Y, ZHENG Y, ZHANG Z. ZmGA3ox2, a candidate gene for a major QTL, qPH3.1, for plant height in maize., 2013, 73(3): 405-416.
[12] YANG N, LU Y, YANG X, HUANG J, ZHOU Y, ALI F, WEN W, LIU J, LI J, YAN J.Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel., 2014, 10(9): e1004573.
[13] PAN Q, XU Y, LI K, PENG Y, ZHAN W, LI W, LI L, YAN J. The genetic basis of plant architecture in 10 maize recombinant inbred line populations., 2017, 175(2): 858-873.
[14] SHAVER G R. Mineral nutrition and leaf longevity inthe role of individual nutrients and the timing of leaf mortality., 1983, 56(2): 160-165.
[15] Dwyer L M, Andrews C J, Stewart D W, MA B L, DUGAS J A. Carbohydrate levels in field-grown leafy and normal maize genotypes., 1995, 35(4): 1020-1027.
[16] Andrews C J, Dwyer L M, Stewart D W, DUGS J A, BONN P, DEYER L M. Distribution of carbohydrate during grainfill in leafy and normal maize hybrids., 2000, 80(1): 87-95.
[17] Stewart D W, Dwyer L M. Mathematical characterization of maize canopies., 1993, 66(3): 247-265.
[18] Modarres A M, Hamilton R I, Dwyer L M, STEWART D W, DIJAK M, SMITH D L. Leafy reduced-stature maize for short-season environments: yield and yield components of inbred lines., 1997, 97(2): 129-138.
[19] Begna S H, Hamilton R I, Dwyer L M, STEWART D W, SMITH D L. Effects of population density on the yield and yield components of leafy reduced-stature maize in short-season areas., 2010, 178(2): 103-110.
[20] DIJAK M, MODARRES A M, HAMILTON R I, DWYER L M, STEWART D W, MATHER D E, SMITH D L. Leafy reduced-stature maize hybrids for short-season environments., 1999, 39(39): 1106-1110.
[21] 曹靖生. 几个玉米株型性状的遗传规律研究. 黑龙江农业科学, 1995(3): 16-19.
CAO J S. Genetic studies on some plant type traits in maize., 1995(3): 16-19. (in Chinese)
[22] CORETEAM R. R: a language and environment for statistical computing.,2015, 14: 12-21.
[23] LI M X, YEUNG J M, CHERNY S S, SHAM P C. Evaluating the effective numbers of independent tests and significant-value thresholds in commercial genotyping arrays and public imputation reference datasets., 2012, 131(5): 747-756.
[24] LI H, PENG Z, YANG X, WANG W, FU J, WANG J H, HAN Y G, CHAI Y C, GUO T T, YANG N, LIU J, WARBURTON M L, CHENG Y B, HAO X M, ZHANG P, ZHAO J Y, LIU Y J, WANG G Y, LI J S, YAN J B. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels., 2013, 45(1): 43-50.
[25] Feng Y, Zheng Q, Song H, WANG Y, WANG A, JIANG L, YAN J, ZHENG Y, YUE B. Multiple loci not only Rf3 involved in the restoration ability of pollen fertility, anther exsertion and pollen shedding to S type cytoplasmic male sterile in maize., 2015, 128(11): 2341-2350.
[26] Yang X, Gao S, Xu S, Zhang Z, Prasanna BM, Li L, Li J, Yan J. Characterization of a global germplasm collection and its potential utilization for analysis of complex quantitative traits in maize., 2011, 28: 511-526
[27] 陈荣江, 刘永录. 玉米若干农艺性状的遗传相关分析. 河南科技学院学报(自然科学版), 1997(2): 19-24.
CHEN R J, LIU Y L. Genetic relation analysis of agronomic characters maize.(), 1997(2): 19-24. (in Chinese)
[28] Xiao Y, Tong H, Yang X, XU S, PAN Q, QIAO F, RAIHAN M S, LUO Y, LIU H, ZHANG X, YANG N, WANG X, DENG M, JIN M, ZHAO L, LUO X, ZHOU Y, LI X, LIU J, ZHAN W, LIU N, WANG H, CHEN G, CAI Y, XU G, WANG W, ZHANG D, YAN J. Genome-wide dissection of the maize ear genetic architecture using multiple populations., 2015, 210(3): 1095-1106.
[29] 郑克志, 李元, 瞿会, 闫伟, 张旷野, 宋茂兴, 吕香玲, 李凤海, 史振声. 玉米株高和穗位高的QTL定位. 江苏农业科学, 2015, 43(5): 61-63.
ZHENG K Z, LI Y, QU H, YAN W, ZHANG K Y, SONG M X, LüX L, LI F H, SHI Z S. QTL mapping of plant height and ear height in maize., 2015, 43(5): 61-63. (in Chinese)
[30] SAMAYOA L F, MALVAR R A, OLUKOLU B A, HOLLAND J B, BUTRON A. Genome-wide association study reveals a set of genes associated with resistance to the mediterranean corn borer (L.) in a maize diversity panel., 2015, 15: 35.
(责任编辑 李莉)
Genome-wide AssociationStudies of Plant Type Traitsin Maize
LIU Kun1, ZHANG XueHai1, SUN GaoYang1, YAN PengShuai1, GUOHaiPing1, CHEN SiYuan2, XUE YaDong1,GUO ZhanYong1, XIEHuiLing1,TANG JiHua1,LI WeiHua1
(1College of Agronomy, HenanAgriculturalUniversity/KeyLaboratory of Wheat and Maize Crops Science,Zhengzhou450002;2No.1 Middle School of Zhengzhou, Zhengzhou450002)
【Objective】Plant morphological traits arethebasis of ideotype-based maize breeding which are closely related to photosyntheticefficiency, lodgingresistance and grain yields. Genome-wide association studies (GWAS) of 558 629 SNPs with genome-wide coverage was used to elucidate the genetic basis of six plant morphological traits, including totalnumber of leaves (LN), leafnumber above ear (LNAN), the ratio of LNAN to LN (LNAN/LN), plantheight (PH), earheight (EH) and the ratio of EHtoPH (EH/PH), which could provide theoretical basis in enhancing ideotype-based maize breeding and facilitating the genetic improvement of new maize varieties with high plant density and lodging resistance. 【Method】Inthisstudy, a representative panel of 284 inbred lines plantedinZhengzhouandXunxian, including temperate,subtropicalandtropicalmaterials, were used for association mapping.【Result】 All traits measured in the two locations exhibited an approximately normal distribution. Highly positive or negative correlations between paired traits were observed. Variance analysis of these traits indicated that significant variations were observed across environment, genotype and the genotype × environment interaction. When test with the optimal GWAS model, we found that Q model showed high type I errors (false positive), while Q+K model were too strict in reducing false positive. K model is the best in reducing false positive. Totally, 56 significant SNP-trait associations involving in 17 loci were identified for five traits (≤3.99E-6), each locus can explain phenotypic variation ranging from 7.97% to 10.56%. Moreover, four loci were detected in both environments, indicating that these 4 loci were less affected by environment effects and could be stable in different environments. All potential candidate genes and their annotations within 100 kb (50 kb upstream and downstream of the lead SNP) of the loci were identified, in total, 80 candidate genes were found, including 42 genes that have functional annotation. For example,the gene GRMZM2G161293 encoding a protein that has acetylgluco-saminyl transferase is associated with plant height and ear height. It catalyzes the transfer of the amino group from N-acetyl glucosamine to glucose, which may improve yield by influencing the content of soluble sugars in maize kernels.【Conclusion】The results indicated that K model having the best result in reducing the type I errors (false positive). Based on K model, a total of 17 loci associated with plant morphological traits were identified.
maize (L.); genome-wide association study; plant type traits; ideotype
2017-09-26;
2017-11-26
国家自然科学基金(U1604231)、河南省重大科技专项(161100110500)、河南省科技攻关项目(152102110060)
刘坤,E-mail:lkun2017@163.com。张雪海,E-mail:xuehai85@126.com。刘坤和张雪海为同等贡献作者。
李卫华,Tel:0371-63558122;E-mail:liwh416@163.com
