肾上腺脑白质营养不良脊髓型一家系报道
2018-03-23张璐璐李建章陈文武
张璐璐 李建章 陈文武
河南大学第一附属医院神经内二科,河南 开封 475001
肾上腺脑白质营养不良脊髓型又称肾上腺脊髓病(adrenomyeloneuropathy,AMN)是肾上腺脑白质营养不良的一特殊的变异类型,为X-连锁的隐形遗传性疾病,是过氧化物酶体病的一种类型。临床上多以成年起病,首先出现的症状为肾上腺皮质功能减退,随后出现进展性的神经功能障碍、括约肌和性功能障碍等,临床少见,容易漏诊、误诊,国内报道多以单个的病例报告多见,本文对作者医院诊断为肾上腺脑白质营养不良脊髓型的1患者进行家系追踪报道(图1)。
1 家系资料
先证者Ⅲ1,患者男性,38岁,以全身皮肤发黑8 a,进行性双下肢无力1 a为主诉入住我医院,患者8 a前逐渐出现全身皮肤发黑,乳晕、皮肤褶皱、面部、牙龈明显发黑,5 a前出现体毛、阴毛、腋毛减少,自己未在意;1 a前出现双下肢发硬,活动不灵活,晨起加重,活动后减轻,后逐渐表现为双下肢无力、僵硬,行走时脚后跟不能着地,双上肢活动及感觉正常,无声音嘶哑、饮水呛咳等症状,发病以来大小便正常,无肌肉萎缩及肌肉跳动等症状,在当地医院诊断为腰椎间盘突出,治疗后效果不佳,随到作者医院,追问病史,患者2 a前既有性功能障碍。既往体健,无糖尿病、冠心病、脑卒中病史,无肝炎、结核等传染病病史,无外伤手术史,无食物药物过敏史,预防接种随当地进行。体格检查:全身皮肤发黑,以面部、乳晕、皮肤褶皱处为著,牙龈明显发黑,巩膜、皮肤无黄染,无肝掌、蜘蛛痣。神经系统检查:高级神经系统检查正常,双侧瞳孔等大等圆,左侧3 mm 右侧3 mm,对光反射存在,双眼活动无异常,闭目有力,双侧软腭上抬有力,咽反射存在,悬雍垂居中,伸舌充分、居中,颈软、无抵抗,双上肢肌力、肌张力正常,双下肢肌张力增高,双下肢肌力4级,痉挛步态,双侧指鼻试验、跟膝胫试验阴性,感觉系统检查正常,双侧Hoffmann征、Babinski征阳,双侧踝阵挛、髌阵挛阳性,脑膜刺激征阴性。血尿粪常规、生化、凝血功能、甲状腺功能、传染病8项无异常,自身免疫性肝炎抗体12项、风湿免疫抗体无异常,脑脊液检查常规、生化、结核、自身免疫性脑炎等未见异常;血ACTH上午8点 :>1 250 pg/mL参考值7.0~61.1 pg/mL;血皮质醇上午8点5.69 μg/dL,参考值7~27 μg/dL;血极长链脂肪酸(VLCFA,C26):1.139 8 pg/mL,高于正常值;C26/C22比值0.83,正常值<0.14。头颅MRI未见异常,胸椎、腰椎MRI无异常,颈椎MRI轻度萎缩,双肾上腺CT:双侧肾上腺萎缩,MEP示:双下肢锥体束传导延迟,双上肢锥体束传导未见异常,SSEP示:双下肢深感觉传导路传导延迟,波幅降低,EMG示:四肢被检肌及神经周围运动及末梢感觉传导未见异常。 入院后考虑诊断肾上腺脑白质营养不良脊髓型,给予补充糖皮质激素、营养神经等药物治疗,皮肤发黑稍有改善,双下肢无力改善不明显。
Ⅲ8 先证者表弟,男30岁,2 a前出现全身皮肤发黑,以乳晕、皮肤褶皱、面部、牙龈处明显,无双下肢无力等症状。神经系统检查:高级神经系统检查正常,脑神经检查无异常,颈软、无抵抗,双上肢肌力、肌张力正常,双下肢肌张力增高,双下肢肌力5级,双侧指鼻试验、跟膝胫试验阴性,感觉系统检查正常,双侧Hoffmann征、Babinski征阳,双侧踝阵挛、髌阵挛阳性,脑膜刺激征阴性。血ACTH上午8点:1 120 pg/mL 参考值7.0~61.1 pg/mL;血皮质醇上午8点6.87 μg/dL,参考值7~27 μg/dL;血极长链脂肪酸(VLCFA,C26):1.127 0 pg/mL;头颅MRI未见异常,颈椎、胸椎、腰椎MRI无异常。
Ⅱ4 先证者舅舅,10 a前因进行性双下肢无力,后长期卧床导致压疮、肺部感染去世,生前皮肤发黑,具体情况不详。
2 讨论
肾上腺功能不全(AI)是肾上腺不能产生足够皮质醇的一种状态[1]。最常见的原因是自身免疫性疾病,占肾上腺功能不全的70%~90%。其余的原因包括感染性肾上腺炎、转移癌、肾上腺出血或梗死、药物和一些遗传性疾病等,如ALD或AMN(表1)[2]。ALD在男性中的发病率估计为1:21 000,女性为1:14 000,在不同种族中无无明显差异[3]。ALD有多种表型,包括儿童脑型、青春期脑型、成人脑型、AMN型、肾上腺皮质功能不全型、橄榄脑桥小脑型、无症状型[4]。由于过氧化物酶体缺乏,导致极长链脂肪酸二十四烷酸(C24:0)和二十六烷酸(C26:0)在组织和体液中聚集,从而引起患者中枢神经系统、周围神经髓鞘、肾上腺损伤[5]。经典的ALD是儿童脑型ALD,通常在4~6岁的童年期发病,伴随着快速进展性的脑脱髓鞘导致痉挛性四肢麻痹、痴呆、癫痫发作、视觉和听觉功能障碍,临床特征也可以在青春期或成年期出现。AMN是ALD的一种特殊类型,是由于Xq-28上的ABCD1基因突变而导致的X连锁的隐性遗传性疾病[5-6]。AMN的发病年龄较晚,通常在30~40岁发病(平均28岁)[7],主要因为脱髓鞘导致脊髓和周围神经损伤,临床表现为逐渐进行性痉挛性截瘫,感觉性共济失调伴振动感觉障碍,括约肌功能障碍,腿痛和阳痿[8]。大约2/3的男性患者有明显或亚临床肾上腺功能不全,大部分患者有相关的性腺功能异常[9]。在我们的家系中,先证者肾上腺功能不全是最初的特征,7 a后出现神经症状。Ⅲ4号患者,最先出现了肾上腺功能不全,无神经系统损害症状,但神经系统检查已出现了上运动神经元损害。神经系统异常可能出现在肾上腺功能不全之前,但高达60%的患者在诊断肾上腺功能不全的时候没有或几乎无神经系统症状[10]。因此,在肾上腺功能不全患者中应考虑临床前AMN。
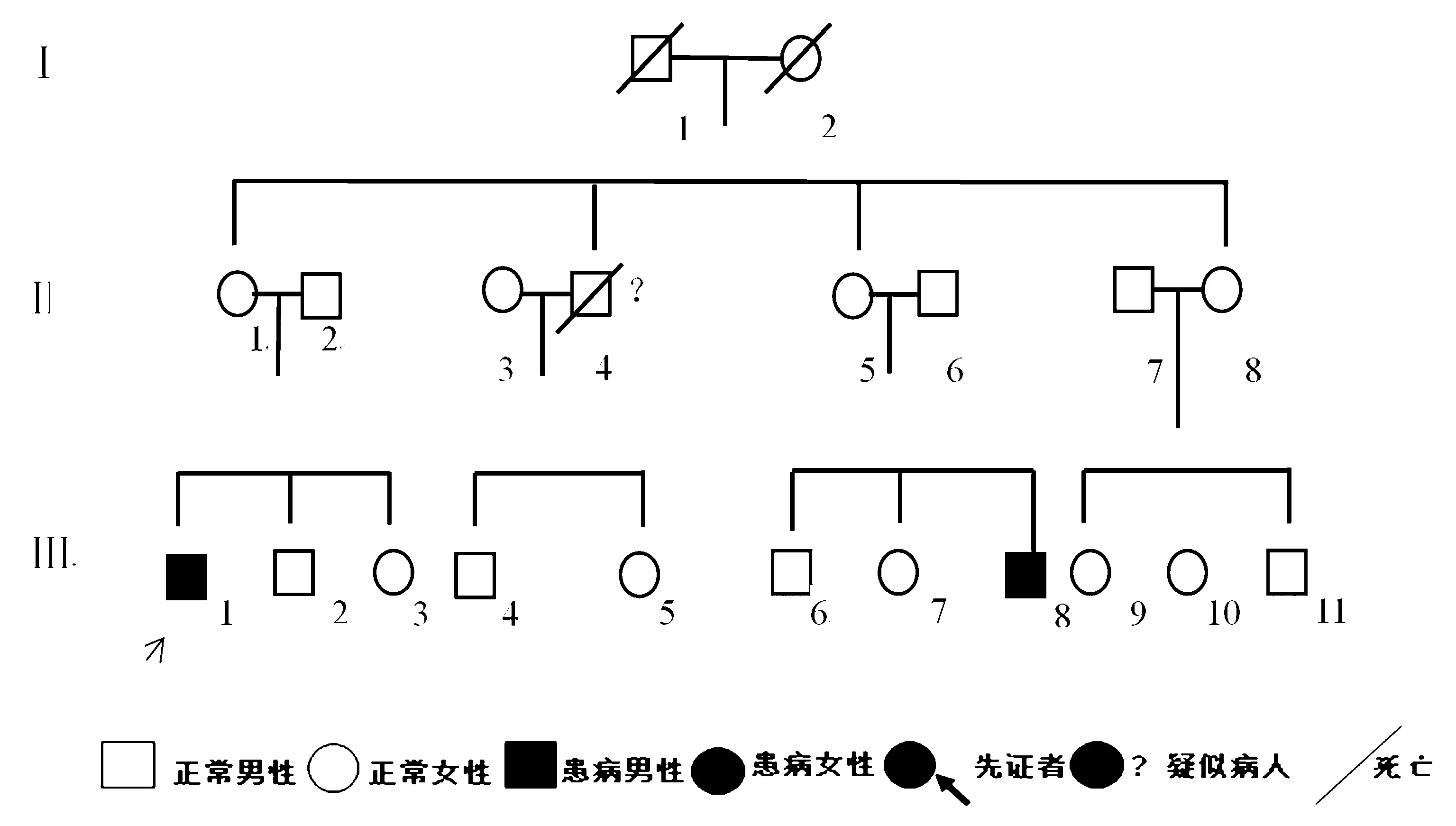
图1 肾上腺脑白质营养不良脊髓型家系图

原因患病率/%自身免疫性肾上腺炎70~90 孤立性肾上腺功能不全 PASⅠ型、Ⅱ型传染性肾上腺炎20 结核 真菌感染 HIV感染 梅毒转移癌(肺癌,乳腺癌,胃癌,结肠癌,淋巴瘤)10肾上腺出血/梗死—药物(酮康唑,氟康唑,利福平,苯妥英)—其他— ALD/AMN 先天性肾上腺发育不良家族性糖皮质激素缺乏/抵抗
PAS:多腺体自体免疫综合征;HIV:人类免疫缺陷病毒;ALD:肾上腺脑白质营养不良;AMN:肾上腺脊髓病
在实验室检查中,VLCFA水平参数已经增加到了三个:二十六烷酸(C26:0),二十六烷酸与二十四烷酸的比值(C26:0/C24:0),和二十六烷酸与二十二烷酸的比值(C26:0/C22:0)。ABCD1基因位点的遗传检测对确诊AMN同样重要。国内王志红等[11]报道一AMN家系,检测到AMN患者血的ABCDl基因第656(G)、657(A)位碱基(位于外显子1)缺失,造成移码突变fsR89。我们的家系经过患者同意后,提取DNA送遗传分子实验室已进行基因突变分析。在电生理研究中,感觉NCS可以是正常的,但通常显示远端潜伏期的轻度延长,幅度减小和传导速度减慢。NCS的异常在男性中比在女性中更常见,下肢比上肢多见,病程长的患者神经传导参数通常比最近诊断出的患者存在更严重的异常[12]。SSEP,特别是下肢可能在早期阶段出现微小的异常,VEP往往是正常的。
国内关于AMN的报道,多以病例报告形式出现,我们的家系中,患者Ⅲ4无神经系统的表现,但已经出现肾上腺功能不全及神经系统的损害,诊断为临床前AMN,在国内尚未报道。所以,临床上仅出现肾上腺功能不全的症状,无神经系统功能损害,不能排除AMN的可能。
[1] BOONEN E,VERVENNE H,MEERSSEMAN P,et al.Reduced cortisol metabolism during critical illness[J].N Engl J Med,2013,368(5):1 477-1 488.
[2] ZELISSEN P M,BAST E J,CROUGHS R J.Associated autoimmunity in Addison’s disease[J].J Autoimmun,1995,8(2):121-130.
[3] ENGELEN M,KEMP S,POLL-THE B T.X-linked adrenoleukodystrophy:pathogenesis and treatment[J].CurrNeurolNeurosci Rep,2014,14(10):486.
[4] MOSER H W,MAHMOOD A,RAYMOND G V.x—linked adrenoleukodystrophy[J].Nat ClinPractNeurol,2007,3(3):140-151.
[5] ROPPER A H,ALLAN H,BROWN R H.The inherited meta-bolic diseasesof the nervous system.In:Ropper AH,Samuels MA,editors.Adams and Victors’ Principles of Neurology[M].8 thed.New York:McGraw-Hill,2005:837-838.
[6] BALCER L J,PRASAD S.Abnormalities of optic nerve and retina.In:Daroff RB,Fenichel GM,Jankovic J,Mazziotta JC,editors.Bradley’s Neurology in Clinical Practice[M].6 thed.Philadelphia:Elsevier Saunders,2012:172-175.
[7] SPUREK M,TAYLOR-GJEVRE R,VAN UUM S,etal.Adrenomyeloneuropathy as a cause of primary adrenal insufficiency and spastic paraparesis[J].CMAJ,2004,171(9):1 073-1 077.
[8] LECUMBERRI B,GIRS M L,COLL M J,et al.Diffuse hair loss in Addison disease:a reason for X-linked adrenoleukodystrophy screening[J].J Am AcadDermatol,2012,66(5):860-861.
[9] ENGELEN M,BARBIER M,DIJKSTRA I M,et al.X-linked adrenoleukodystrophy in women:a cross-sectional cohort study[J].Brain,2014,137(3):693-706.
[10] SADEGHI-NEJAD A,SENIOR B.Adrenomyeloneuropathy presenting as Addison’s disease in childhood[J].N Engl J Med,1990,322(3):13-16.
[11] 王志红.一个肾上腺脑白质营养不良家系的ABCDl基因突变分析[J].中华神经医学杂志,2010,9(10):1 045-1 047.
[12] CHAUDHRY V,MOSER H W,CORNBLATH D R.Nerve conduction studies in adrenomyeloneuropathy[J].J Neurol Neurosurg Psychiatry,1996,61(7):181-185.
