里氏木霉组成型表达siRNA干扰cre1基因对纤维素酶表达的调控作用
2018-03-21高云雨钟路遥董冠园佘伟怡周娇娇刘思远田生礼
高云雨, 钟路遥, 董冠园, 佘伟怡, 周娇娇, 刘思远, 田生礼
(深圳大学生命与海洋科学学院 深圳市微生物基因工程重点实验室,广东 深圳 518052)
里氏木霉(Trichodermareesei)是一种丝状真菌,是重要的纤维素酶产生菌,广泛用于生产纤维素酶[1]。里氏木霉纤维素酶是一个复杂的酶体系,由三类酶组成,包括内切葡聚糖酶(endo-β-1,4-D-glucanase,EG)、外切葡聚糖酶(exo-1,4-β-D-g1ucanase,CBH)和 β-葡萄糖苷酶(β-g1ucosidase,BGL)。里氏木霉具有强大的合成和分泌蛋白的能力,其主要生产纤维素酶和纤维二糖水解酶 I (CBH1),表达量最高的菌株分泌的纤维素酶总量可达 100 g/L[2]。里氏木霉的基因组只有33.9 Mb,比其他丝状真菌的要小得多[3]。而且其安全无毒易于培养,因此,里氏木霉是重组蛋白表达和生产的优良宿主,通过基因工程等手段可以高效调控里氏木霉纤维素酶基因以提高纤维素酶产量。里氏木霉纤维素酶基因的表达存在葡萄糖效应,当存在快速利用碳源如葡萄糖时纤维素酶基因的表达会受抑制,这是因为葡萄糖诱导了里氏木霉中碳源阻遏调控因子的表达,目前公开发表的两个阻遏蛋白是CRE1和ACE1。CRE1蛋白是Cys2-His2型转录因子[4],是一个广域转录抑制因子,能调节纤维素酶和木聚糖酶基因的表达,参与脯氨酸代谢、乙醇代谢、多糖水解等过程中的基因转录调控[5]。研究表明,敲除或抑制里氏木霉野生型菌株中的cre1基因,可显著提高纤维素酶的表达[6-7]。但是完全敲除cre1会引起纤维素酶以外的其他基因的表达变化,所以直接敲除cre1基因不是提高纤维素酶表达的最佳方法[8],而采用RNAi对cre1基因进行抑制的方法可以抑制cre1的表达同时对其他无关基因不产生影响。ACE1是一个含有3个Cys2-His2类锌指结构的DNA结合蛋白,ACE1为转录抑制因子,在里氏木霉中敲除基因ace1后,在纤维素和槐糖的培养条件下能够提高主要的纤维素酶基因和半纤维素酶基因表达量[9]。目前,RNA干扰技术(RNA Interference, RNAi)在真菌中应用广泛,应用RNAi技术对丝状真菌如康氏木霉、里氏木霉和嗜热毁丝霉的cre1基因的调控作用已有较多的研究发表[7,10-11]。大部分使用的是长链RNAi干扰技术以及诱导型干扰载体,对cre1使用短链siRNA组成型干扰载体未见报道。本文将利用Wang等[12]从里氏木霉糖代谢基因中筛选出了丙酮酸脱羧酶基因(pyruvate decarboxylase,pdc)的组成型强启动子Ppdc和终止子Tpdc构建里该氏木霉组成型表达载体。李洁璇等[13]已经在里氏木霉中应用该启动子和终止子构建了红色荧光蛋白的siRNA干扰载体,并且成功地在里氏木霉中干扰了红色荧光蛋白的表达,但其使用组成型siRNA干扰载体仍然需要和PAN7-1共转化以获得真菌的潮霉素抗性。本研究通过构建里氏木霉cre1组成型siRNA干扰载体,使用组成型启动子和终止子同时包含潮霉素抗性基因,可高效便捷地干扰里氏木霉cer1基因的表达,探讨里氏木霉cre1基因沉默后,对纤维素酶基因的表达会如何变化,为里氏木霉纤维素酶基因的多靶向表达调控提供新的思路和实验方法。
1 材料与方法
1.1 材料
1.1.1 菌株及质粒 大肠埃希菌JM107由本实验室保存,里氏木霉QM9414购自美国模式菌种收藏中心(ATCC);质粒pLXT带有真菌筛选标记潮霉素B抗性基因(Hph)和大肠埃希菌筛选标记氨苄青霉素抗性基因(Amp),由本实验室构建保存。
1.1.2 培养基 里氏木霉液体基本培养基:KH2PO42 g/L,(NH4)2SO41.4 g/L,尿素0.3 g/L,CaCl2·2H2O 0.4 g/L,FeSO4·7H2O 0.005 g/L,MnSO4·H2O 0.001 6 g/L,ZnSO4·7H2O 0.001 7 g/L,CoCl2·6H2O 0.003 7 g/L,蛋白胨2 g/L,吐温-80 1 g/L,葡萄糖20 g/L。
1.1.3 试剂 各种DNA工具酶、T4 DNA连接酶、DNA Marker和RNase Free H2O等均购自TaKaRa 公司;DNA胶回收纯化试剂盒、PCR产物回收纯化试剂盒购自上海生工生物工程有限公司;真菌总RNA提取试剂盒购自Omega公司;反转录试剂盒(EasyScript One-Step gDNA Removal and cDNA Synthesis SuperMix)购自北京全式金生物技术有限公司;qPCR试剂盒(Bestar®SybrGreen qPCR mastermix)购自DBI Bioscience公司;溶壁酶(Lysing Enzyme from Trichoderma harzianum)购自Sigma公司;潮霉素和氨苄青霉购自Invitrogen公司;其他试剂均购自上海生工生物工程有限公司。
1.1.4 仪器 高压自动灭菌锅(Hirayama,HVE-50);超净工作台(苏州净化设备总厂,SW-CJID);荧光定量PCR仪(analytik jena, qTOWER3);PCR仪(ABI,2720);冷冻离心机(Thermo Scientific,Multifuge XIR);NanoDrop2000超微量分光光度计(Thermo Scientific,ND2000);小型离心机(Eppendorf,Minispin);pH计(Mettler-Toledo,320-S);电泳仪(Amersham Pharmacia,EPS-601);水平电泳槽(北京六一,DYCP-31D);高纯水仪(力新仪器有限公司,Heal force ROP3);核酸成像系统(Bio-Rad,GEL DOX XR+);低温摇床(上海苏坤,SKY-200B);低温培养箱(上海爱朗,LTI-700);奥林匹斯BX51TRF System (东京,日本)。
1.2 方法
1.2.1cre1干扰片段的设计与合成 根据GenBank公布的CRE1的Protein ID:120117,在里氏木霉基因组网站(http://genome.jgi-psf.org/Trire2/Trire2.home.html)查找到cre1基因全序列。cre1位于scaffold 2中785 322~788 378 bp,包含1个外显子,基因长度为3 075 bp。将cre1基因的mRNA序列,输入网站(Invitrogen Block-iT RNAi Designer)进行siRNA靶序列设计,选择其中3条作用于不同位点的siRNA干扰片段进行实验,根据靶序列的位置顺序命名为cre1-T4、cre1-T7和cre1-T10。再另选一条将其序列顺序打乱,作为阴性对照,命名为cre1-Neg。设计合成干扰片段的序列见表1。cre1的siRNA干扰片段分别由正、反义siRNA序列构成,中间使用9 nt(TTCAAGAGA)的稳定loop作为茎环连接siRNA,同时在该片段两端5′端加入一个突出dA,便于与所构建的T载体pLXT连接。分别设计好siRNA的正义链和反义链,由Invitrogen公司合成寡核苷酸链。然后把待退火的DNA oligos分别用双蒸水配制成50 μmol/L浓度,退火后获得的双链DNA长度为54 bp。

表1 cre1的siRNA干扰片段
1.2.2 干扰表达盒的构建 该载体的一个特征是含有潮霉素抗性基因,只需转化一个DNA片段即可获得含有抗性基因的阳性转化子,减少筛选的工作量。含潮霉素抗性基因的干扰载体pLXT(载体图谱见图1),通过限制性内切酶AhdI酶切后,载体DNA双链的3′端都悬挂一个胸腺嘧啶残基dT,设计的干扰片段5′端都带有一个dA,所以载体可直接连接干扰片段,以期实现对表达宿主里氏木霉高效转化和对靶基因的干扰表达。构建成功的含有干扰cre1基因的重组体,提取质粒分别命名为Ppdc-T4-Tpdc、Ppdc-T7-Tpdc、Ppdc-T10-Tpdc和Ppdc-Neg-Tpdc。由于载体上有潮霉素抗性基因hph,质粒可直接进行里氏木霉原生质体转化。

图1 载体pLXT图谱Fig.1 A plasmid map of pLXT vector
1.2.4 基因组DNA提取 将PCR筛选出来的转化子接种于PDA平板上,28 ℃恒温培养7 d后,制备适量孢子悬液接种于液体基本培养基中,28 ℃、250 r/min培养2 d。抽滤菌液,得到菌丝体,液氮研磨成粉后使用omega的E.Z.N.A® Fungal DNA Kit提取基因组DNA。用NanoDrop2000超微量分光光度计和琼脂糖凝胶电泳检测DNA样品的浓度和质量。
1.2.5 RNA提取 为分析siRNA干扰片段对纤维素酶活力的影响,将重组菌T.reesei-cre1-T4、T.reesei-cre1-T7、T.reesei-cre1-T10、T.reesei-cre1-Neg和出发菌株T.reeseiQM9414分别接种于10 mL基本培养基中,每个菌株分别做3个平行样品,28 ℃、250 r/min培养48 h后,每种重组菌取1.5 mL菌液接种到30 mL含有0.1%微晶纤维素(质量分数)的产酶培养基中,28 ℃、250 r/min培养48 h和144 h后,利用omega的E.Z.N.A®. Fungal RNA Kit试剂盒,按照说明书的方法将菌丝使用液氮研磨成粉后提取总RNA。用NanoDrop2000超微量分光光度计和琼脂糖凝胶电泳检测RNA样品的浓度和质量。
1.2.6 RNA反转录和荧光定量PCR 使用反转录试剂盒,以Random Primer为引物,将约100 ng RNA反转录成cDNA。使用荧光定量 PCR 仪进行荧光定量PCR。每个反应20 μL体积,包括2 μL模板(1∶ 60稀释反转录产物),10 μL 2×SybrGreen qPCR Master Mix,8 μmol/L 正反向引物(表2)各0.5 μL和7 μL nuclease-free水。程序设置:预变性95 ℃ 30 s,95 ℃变性5 s,60 ℃退火延伸31 s,运行40个循环。运行结束后得到融解曲线,用以核对PCR产物的特异性。所有PCR反应每个样品做3个复孔。目的基因的表达量通过内参基因sar1校正。以对照菌株的表达量为1,其他转化子的表达量与对照样品的比值作为数据分析。

表2 荧光定量PCR使用的引物
1.2.7 酶活测定 为分析siRNA干扰片段对纤维素酶活力的影响,将重组菌T.reesei-cre1-T4、T.reesei-cre1-T7、T.reesei-cre1-T10、T.reesei-cre1-Neg和出发菌株T.reeseiQM9414分别接种于30 mL基本培养基中,每个菌株分别做3个平行样品,28 ℃、250 r/min培养48 h后,每种重组菌取1.5 mL菌液接种到30 mL含有0.1%(质量分数)微晶纤维素的产酶培养基中,28 ℃、250 r/min培养,一般来说,里氏木霉的滤纸酶活性在诱导培养基中会随着诱导时间的延长而增加,并将在诱导约144 h达到最大[15]。所以在48 h和144 h分别收集酶液,将粗酶液稀释一定倍数后测定其滤纸酶活和CMC酶活。滤纸酶活(FPA)和羧甲基纤维素(CMC)酶活测定方法参考Ghose[16]。以CMC和Whatman No.1滤纸为底物,分别加酶液0.5 mL, 50 ℃孵育30 min和1 h,加DNS沸水浴5 min。测定OD540值,每个样品做3个重复,结果取平均值。以1 h内水解50 mg滤纸释放出2.0 mg葡萄糖或30 min内水解2%羧甲基纤维素释放出0.5 mg葡萄糖的酶液稀释倍数来计算FPU和CMCase活性单位,单位为IU/mL。
2 结果与分析
2.1 里氏木霉siRNAW干扰表达盒完整性检测
将重组菌T.reesei-cre1-T4、T.reesei-cre1-T7、T.reesei-cre1-T10、T.reesei-cre1-Neg和出发菌株T.reeseiQM9414分别提取转化子基因组进行PCR鉴定,使用引物1:5′-CCTGCAAGTCTCCATCACAAG-3′,引物2:5′-TTCATAGTCCCATTGTCAGCA-3′,结果见图2。结果显示在1 000 bp处有明显条带,该条带的大小与siRNA干扰表达盒的大小基本一致,进一步对PCR扩增的DNA片段进行测序分析,表明DNA序列与siRNA干扰表达盒的序列一致,进一步证明了干扰表达盒成功整合到里氏木霉QM9414基因组中。
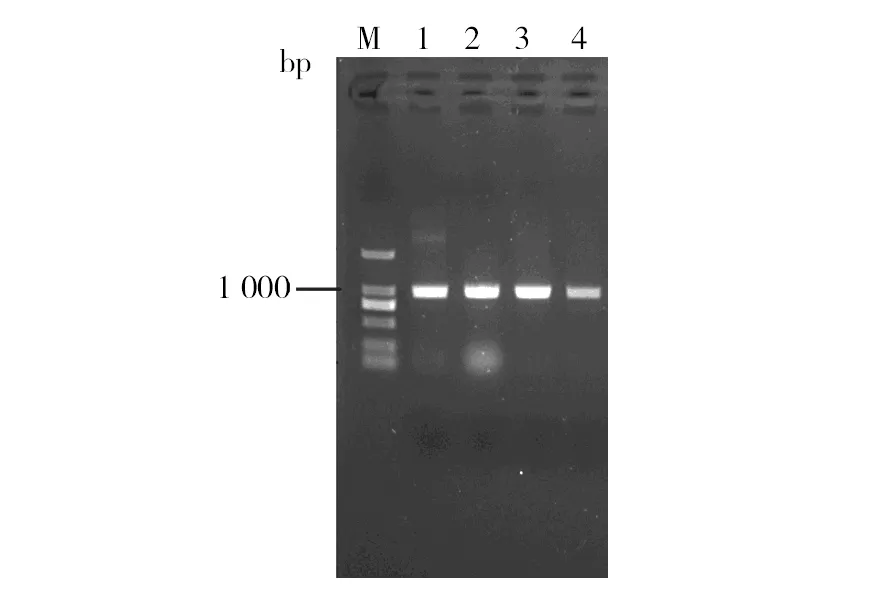
图2 PCR鉴定里氏木霉基因组中的 siRNA干扰表达盒Fig.2 PCR amplified siRNA disruption cassettes from recombinant strains M:Takara DL 2 000 DNA Marker;1~4:分别从重组菌T. reesei-cre1-T4、T. reesei-cre1-T7、T. reesei-cre1-T10和T. reesei-cre1-Neg的基因组中扩增出来的siRNA干扰表达盒 M: Takara DL 2 000 DNA Marker;1-4:The PCR amplified siRNA disruption cassettes from recombinant strains T.reesei-cre1-T4,T.reesei-cre1-T7,T.reesei-cre1-T10 and T.reesei-cre1-Neg
2.2 干扰重组菌的纤维素酶酶活的测定
滤纸酶活代表的是纤维素酶三种酶组分,即内切型葡聚糖酶、外切型葡聚糖酶、β-葡聚糖苷酶协同作用后的总酶活,是菌株整个纤维素酶系酶活力水平的综合体现。重组菌在产酶培养基中48 h和144 h的滤纸酶活如图3所示,根据实验结果,所有菌株在诱导培养约144 h时滤纸酶活均比48 h时高。其中,含有siRNA干扰片段的重组菌T.reesei-cre1-T4、T.reesei-cre1-T7和T.reesei-cre1-T10的滤纸酶活均是T.reesei-cre1-Neg和出发菌株T.reeseiQM9414的1倍以上,而阴性对照和出发菌株的酶活力相近。
外切葡聚糖酶对纤维素链具有高度的专一性,而内切葡聚糖酶的专一性较低,所以以羟甲基纤维素为底物时测得的酶活(CMCase)主要反映内切β-1,4-葡聚糖苷酶的活力,结果见图 4。其中,含有siRNA干扰片段的重组菌株T.reesei-cre1-T4、T.reesei-cre1-T7和T.reesei-cre1-T10的CMC酶活都是出发菌株T.reeseiQM9414的1.8倍以上。T.reesei-cre1-T10的CMC酶活力最高可达7.2 IU/mL。出发菌株T.reeseiQM9414和阴性对照菌株T.reesei-cre4-Neg的酶活也在144 h有明显增长趋势,但两者的酶活力均低于干扰重组菌。

图3 重组菌在48 h和144 h时产酶培养基中分泌 的滤纸酶活Fig.3 The FPA produced by recombinant strains in induction medium of 48 h and 144 h T. reesei-cre1-T4、T. reesei-cre1-T7和T. reesei-cre1-T10:cre1的siRNA干扰重组菌;T. reesei-cre1-Neg:干扰重组菌的阴性对照菌株;T. reesei QM9414:里氏木霉原菌。*P<0.05, **P<0.01,图4~9同 T. reesei-cre1-T4, T. reesei-cre1-T7 and T. reesei-cre1-T10:The siRNA recombinant strain of cre1; T. reesei-cre1-Neg:The negative control of the recombinant strain; T. reesei QM9414:The starting strain. *P<0.05, **P<0.01,the same as figure 4-9

图4 重组菌在48 h和144 h时产酶培养基中分泌的CMC酶活Fig.4 The CMCase produced by recombinant strains in induction medium of 48 h and 144 h
2.3 干扰重组菌转录水平的cre1和ace1相对表达量分析
对所有样品进行实时荧光定量PCR反应,使用qPCR Software 3.2软件对实验结果进行处理分析。实验数据经内参基因sar1定量值校正后,分别以出发菌株T.reeseiQM9414中cre1和ace1的表达量为1,计算出培养48 h和144 h的干扰重组菌中cre1和ace1的相对表达量,结果见图5、图6。
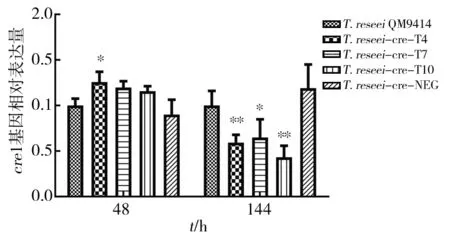
图5 干扰重组菌分别诱导培养48 h 和144 h cre1的相对表达量Fig.5 cre1 gene relative expression level of recombinant strains after respectively inducing 48 h and 144 h
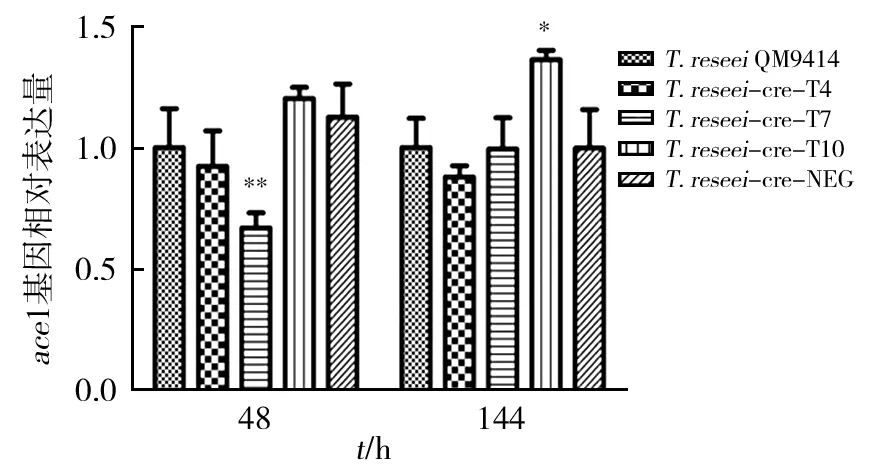
图6 干扰重组菌分别诱导培养48 h 和144 h ace1的相对表达量Fig.6 ace1 gene relative expression level of recombinant strains after respectively inducing 48 h and 144 h
结果显示,在诱导培养48 h时,干扰重组菌T.reesei-cre1-T4、T.reesei-cre1-T7、T.reesei-cre1-T10的cre1 mRNA表达量与出发菌株T.reeseiQM9414相比,分别约为出发菌株的1.3、1.2和1.5倍。然而,在诱导培养144 h后,T.reesei-cre1-T4、T.reesei-cre1-T7、T.reesei-cre1-T10的cre1 mRNA表达量是出发菌株T.reeseiQM9414的0.5、0.6和0.4倍。而含有无同源序列的阴性对照菌株T.reesei-cre1-Neg中cre1的表达量与出发菌相近,结果表明对cre1基因进行siRNA干扰的效果明显。ACE1 和CRE1同样是阻遏蛋白,研究表明,ACE1是负调节因子,ace1基因的缺失会导致cbh1、cbh2、egl1和egl2基因的转录增加2~30倍,其与cre1基因的调节机制是不相关的,是相互独立的调节机制[9]。本研究对ace1基因的转录水平进行qPCR 验证,结果见图6,结果表明对cre1的siRNA干扰没有引起ace1的表达变化,证实了cre1的干扰表达不影响ace1的表达,纤维素酶活力变化的原因不是由于ace1基因的影响。
纤维素酶是起协同作用的多组分酶系,不仅具有内切葡聚糖酶和外切葡聚糖酶活性,还有很高活力的木聚糖酶。cbh1是纤维二糖水解酶CBH1的编码基因,egl1是内切β-1,4-葡聚糖酶EGL1的编码基因[17]。文献报道,XYR1是主要纤维素酶和半纤维素酶基因包括cbh1、cbh2、egl1、bgl1、xyn1和xyn2等转录所必须的激活因子[18]。作为上述基因的转录激活因子,研究显示xyr1自身的转录受到cre1和ace1的调控[17-19]。为验证干扰cre1是否会有效解除对纤维素酶基因或其激活因子的抑制作用,分别使用荧光定量PCR分析干扰重组菌中纤维素酶基因cbh1、egl1和木聚糖酶激活因子XRY1的相对表达量。
cbh1主要编码纤维二糖水解酶CBH1。对干扰重组菌进行荧光定量PCR分析,结果见图7。在诱导48 h时,重组菌cbh1的表达量与出发菌株相差不大,但是在诱导144 h后分别是同时段出发菌QM9414的1.9、2.5和2.5倍。说明cre1对激活蛋白CBH1编码基因的转录具有激活作用。
egl1是内切β-1,4-葡聚糖酶Egl1的编码基因。通过荧光定量PCR手段,分析重组菌中egl1的相对表达量,结果见图8。在诱导144 h时,重组菌T.reesei-cre1-T4、T.reesei-cre1-T7、T.reesei-cre1-T10中egl1的相对表达量分别是QM9414的2.7、2.5和2.9倍。由此可知,在cre1受到有效干扰的菌株中,内切β-1,4-葡聚糖酶的表达可能由于解除了抑制而高效表达。说明抑制cre1的表达不仅部分解除纤维素酶合成受葡萄糖分解代谢物阻遏作用,而且激活纤维素酶中egl1基因的转录。
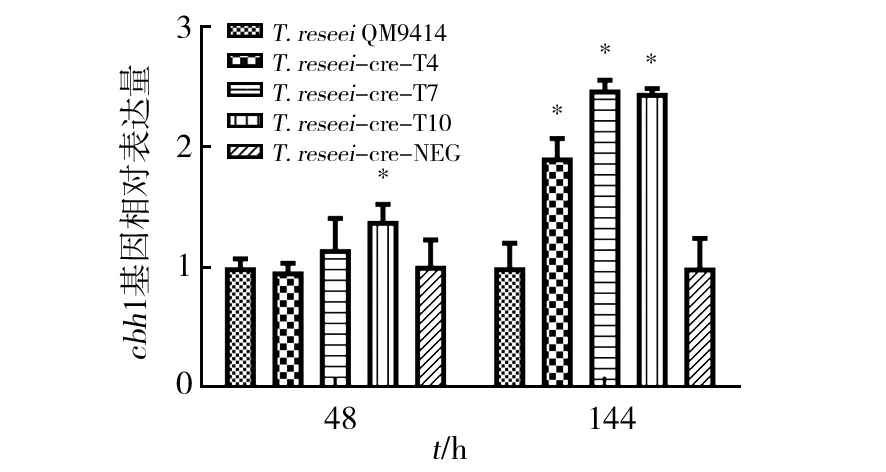
图7 干扰重组菌分别诱导培养48 h和 144 h cbh1的相对表达量Fig.7 cbh1 gene relative expression level of recombinant strains after respectively inducing 48 h and 144 h
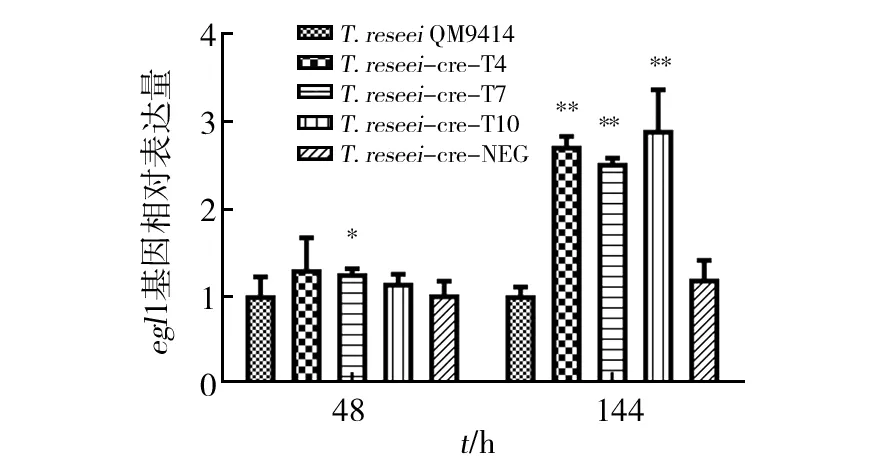
图8 干扰重组菌分别诱导培养48 h和 144 h egl1的相对表达量Fig.8 egl1 gene relative expression level of recombinant strains after respectively inducing 48 h and 144 h
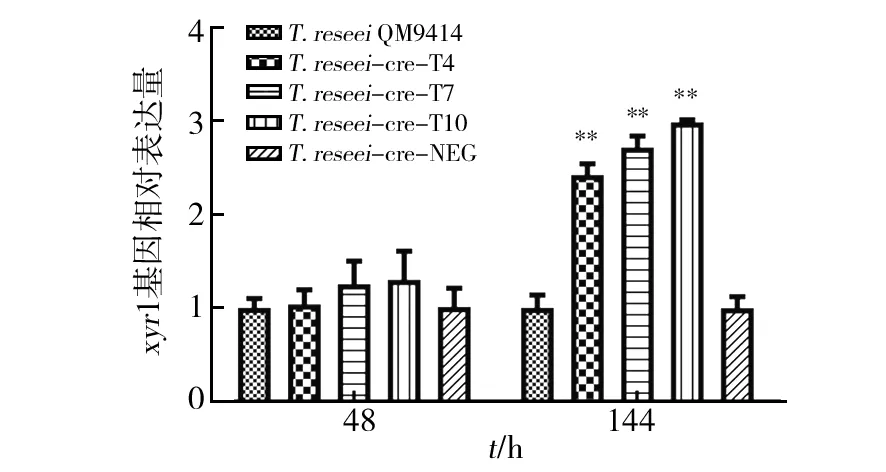
图9 干扰重组菌分别诱导培养48 h和 144 h xyr1的相对表达量Fig.9 Gene xyr1 relative expression level of recombinant strains after respectively inducing 48 h and 144 h
xry1是木聚糖酶激活因子XRY1的编码基因。因此通过荧光定量PCR手段,分析重组菌中xyr1的相对表达量,结果见图9。在诱导培养基中培养48 h时,干扰重组菌T.reesei-cre1-T4、T.reesei-cre1-T7、T.reesei-cre1-T10的xyr1表达量分别与原菌相差不大,但在诱导144 h后发现,此时干扰重组菌T.reesei-cre1-T4、T.reesei-cre1-T7、T.reesei-cre1-T10的xyr1相对表达量分别是同时期T.reeseiQM9414中xyr1基因表达量的2.4、2.7和2.8倍,阴性对照菌株和T.reesei-cre1-Neg和出发菌株中xyr1表达量均变化不大。这说明xyr1基因在干扰cre1基因的转录后获得高效转录。
3 讨 论
里氏木霉是工业生产纤维素酶的重要菌种。由于其存在的“碳抑制效应”,所以在碳源丰富的条件下,纤维素酶的合成受到分解代谢阻遏物的抑制[20-21]。为提高里氏木霉中纤维素酶的产量,需要从纤维素酶表达调控系统入手进行研究。至今对里氏木霉纤维素酶表达机制的研究尚不深入。本研究就里氏木霉碳阻遏抑制因子cre1对里氏木霉产纤维素酶的影响进行了研究。目前使用siRNA对里氏木霉进行干扰研究的报道较少,一些基因如果直接被敲除会引发一系列的反应甚至致死[22],而通过RNA干扰技术则可以降低基因表达量的同时避免这些问题。本研究构建siRNA干扰载体,使用特异性小片段连接组成型启动子和终止子,不受诱导物的影响,可高效稳定地表达干扰基因,直接合成的小片段还可以随时更换为其他基因,为里氏木霉其他基因甚至多基因的干扰研究提供极大的便利。该载体还包含潮霉素B抗性基因,而且原生质体转化过程中省略了包含潮霉素B抗性基因的载体pAN7-1的提取和共转化步骤,不仅大大减少了工作量,并可提高转化效率。
通过表达siRNA干扰载体,实现了对cre1基因的干扰抑制,而其他阻遏因子ace1的基因表达则不受影响。两种酶活性测定的结果显示,转入不同干扰片段重组菌中的滤纸酶活和CMC酶活均高于出发菌株T.reeseiQM9414的酶活性水平,说明酶活性的提高与干扰cre1有关,酶活性提高的原因可能是由于cre1的干扰导致分解代谢阻遏总体效应的下降,从而解除了阻遏物对纤维素酶基因合成的抑制,菌株在短期内大量分泌纤维素酶。酶活性从一定程度上证明了通过RNA干扰cre1基因,降低了cre1的转录表达调控,进一步对纤维素酶基因cbh1、egl1及木聚糖酶激活因子XYR1的mRNA表达水平进行了荧光定量PCR检测,结果显示,与出发菌相比,cre1干扰重组菌中的cbh1、egl1和xyr1的表达均有明显提高,说明干扰cre1可以解除阻遏物对部分纤维素酶基因表达的抑制,解除cre1的抑制对纤维素酶及其相关基因的表达和调控都具有重要意义。
本研究通过纤维素酶酶活和mRNA水平等分析,进一步确定了沉默cre1基因可提高里氏木霉中相关纤维素酶的产量。从而也证实了通过组成型siRNA干扰载体可以对里氏木霉进行方便、高效和快捷的基因调控,为进一步对里氏木霉多靶向基因调控等提供参考。
[1] Mach RL, Strauss J, Zeilinger S, et al. Carbon catabolite repression of xylanase I (xyn1) gene expression inTrichodermareesei[J].Molecular Microbiology,1996,21(6):1273-1281.
[2] Zhang GT, Hartl L, Schuster A, et al. Gene targeting in a nonhomologous end joining deficientHypocreajecorinai[J]. Journal of Biotechnology, 2009, 139(2): 146-151.
[3] Martinez D, Berka R M, Henrissat B, et al. Genome sequencing and analysis of the biomass-degrading fungusTrichodermareesei(syn.Hypocreajecorina)[J].Nature Biotechnology, 2008, 26(5): 553-560.
[5] Strauss J, Mach RL, Zeilinger S, et al. CRE1, the carbon catabolite repressor protein fromTrichodermareesei[J]. FEBS Letters, 1995, 376 (1-2): 103-107.
[7] Wang SW, Liu G, Yu J, et al. RNA interference with carbon catabolite repression inTrichodermakoningiifor enhancing cellulase production[J]. Enzyme & Microbial Technology, 2013, 53(2):104-109.
[8] Portnoy T, Margeot A, Seidl-Seiboth V, et al. Differential regulation of the cellulase transcription factors XYR1, ACE2, and ACE1 in Trichoderma reesei strains producing high and low levels of cellulase[J]. Eukaryotic Cell, 2011, 10(2):262-271.
[9] Aro N, Ilmén M, Saloheimo A, et al. ACEI ofTrichodermareeseiis a repressor of cellulase and xylanase expression[J]. Applied & Environmental Microbiology, 2003, 69(1):56-65.
[10] 王榕,宫莉,姚斌,等.利用RNAi抑制cre1基因转录提高里氏木霉表达纤维素酶[J].食品工业科技, 2016,11:189-194.
[11] Yang F, Gong YF, Liu G, et al. Enhancing cellulase production in thermophilic fungusMyceliophthorathermophilaATCC42464 by RNA interference ofcre1 gene expression[J]. Journal of Microbiology and Biotechnology,2015,25(7): 1101-1107.
[12] Wang SW, Liu G, Wang J, et al. Enhancing cellulase production inTrichodermareeseiRUT C30 through combined manipulation of activating and repressing genes[J]. Journal of Industrial Microbiology and Biotechnology, 2013, 40(6): 633-641.
[13] 李洁璇,刘雯莉,梁智成,等. 丝状真菌里氏木霉中shRNA沉默红色荧光基因[J]. 微生物学杂志,2016,36(4):7-15.
[15] Sternberg D, Mandels GR. Regulation of the cellulolytic system inTrichodermareeseiby sophorose: induction of cellulase and repression of β-glucosidase[J]. Journal of Bacteriology, 1981, 144 (3): 1197-1199.
[16] Ghose TK. Measurement of cellulase activities[J]. Pure and Applied Chemistry, 1987,59(2):257-268.
[17] Aro N, Saloheimo A, Ilmén M, et al. ACEII, a novel transcriptional activator involved in regulation of cellulase and xylanase genes ofTrichodermareesei[J].Journal of Biological Chemistry, 2001, 276 (26): 24309-24314.
[18] Stricker AR, Mach RL, de Graaff L H. Regulation of transcription of cellulases- and hemicellulases-encoding genes inAspergillusnigerandHypocreajecorina(Trichodermareesei) [J]. Applied Microbiology and Biotechnology, 2008, 78 (2): 211-220.
[19] Saloheimo A, Aro N, Ilmén M, et al. Isolation of theace1 gene encoding a Cys2-His2transcription factor involved in regulation of activity of the cellulase promotercbh1 ofTrichodermareesei[J]. Journal of Biological Chemistry, 2000, 275(8): 5817-5825.
[20] Portnoy T, Margeot A, Linke R, et al. The CRE1 carbon catabolite repressor of the fungusTrichodermareesei: a master regulator of carbon assimilation[J]. BioMed Central Genomics, 2011, 12(1): 1-12.
[21] Lockington RA, Kelly JM. The WD40-repeat protein CREC interacts with and stabilizes the deubiquitinating enzyme CREB in vivo inAspergillusnidulans[J]. Molecular Microbiology, 2002,43(5):1173-1182.
[22] Goldoni M, Azzalin G, Macino G, et al. Efficient gene silencing by expression of double stranded RNA in Neurospora crassa[J].Fungal Genet Biol, 2004, 41(11):1016-1024.
