流速对nTiO2和磷酸盐迁移的影响及模型分析
2018-03-21李祖玲曹玉和孙可青
冯 刚,徐 楠,李祖玲,曹玉和,孙可青
(苏州科技大学 化学生物与材料工程学院,江苏 苏州 215009;江苏省环境功能材料重点实验室,江苏 苏州 215009)
纳米二氧化钛(nTiO2)是大量生产的金属氧化物纳米材料之一,由于纳米级材料表现出的超高的光催化能力,已经越来越多的被应用于各种领域和商业产品[1-5]。在大量生产及广泛应用中,一些nTiO2无法避免地释放到自然水体与土壤环境中。大量证据表明:人工nTiO2进入水体后,对水生生物有着不利的影响,这些水生生物包括微生物、藻类、无脊椎动物和鱼类[6-7]。因此,研究附着在土壤饱和颗粒上的纳米微粒和存在于土壤颗粒饱和孔隙介质中的纳米微粒之间的相互关系,已经成为纳米颗粒环境行为研究的热点,目前国际上研究的人工纳米材料主要是工业富勒烯nC60、碳纳米管和二氧化硅纳米微粒,这些纳米材料在饱和多孔介质中的迁移性各有差异,溶液的流速、离子浓度以及纳米微粒的表面电位都会影响其迁移行为[8-9],所以有必要探索nTiO2在不同环境条件下表面性能的变化以及在自然界中的迁移情况。在农业生产以及人们的生活生产中,含磷物质的大量使用,使得含磷物质进入到土壤和水体中。nTiO2颗粒对磷酸盐有一定的吸附作用,使得其表面性质发生变化,最终导致nTiO2在土壤中的迁移性质发生变化[10-11]。因此,该文将探索nTiO2在不同环境条件下表面Zeta电位的变化以及在磷酸盐环境下不同环境水流速度对其迁移的影响。
1 材料与方法
1.1 nTiO2磷酸盐悬浮液的制备
实验所用的所有化学药品均为分析纯,购置于国药集团。20 nm nTiO2购自上海高全化工有限公司,称取1 g·L-1nTiO2溶于 0.1 mM 磷酸盐溶液(NaH2PO4)和 10 mM NaNO3电解质溶液中,用超声波清洗器(KQ 2200B,Ultrasonic Instruments Co.,Ltd.,Hunshan,China)超声 30 min,用于迁移实验。
1.2 nTiO2颗粒表面Zeta电位的测试
准确称取0.01 g nTiO2颗粒于100 mL烧杯中,配制成不同条件的悬浮液。不同条件分别为电解质NaNO3浓度(0.1-5 mM)和磷酸盐溶液(0.1-5 mM),其中所有的悬浮液pH值用稀释的HCl和NaOH调节到pH=6.50。然后,将调节好pH值的nTiO2颗粒的悬浮液放置在超声仪器上超声30 min。超声结束后,将悬浮液置于搅拌器上搅拌30 s。最后,用马尔文Nano-ZS90测试nTiO2颗粒的Zeta电位,所有样品测试三次,取平均值作为最终的Zeta电位值。
1.3 石英砂柱迁移实验
参考Fang等研究方法[2],采用柱淋溶实验研究nTiO2的迁移行为。选用长17.5 cm、内径25 mm的层析柱。用石英砂填充层析柱,并用去离子水饱和12 h。用蠕动泵向已经饱和过的层析柱注入200 mL(10 mM NaNO3)背景溶液,用自动样品部分收集器(BS-100A,上海沪西)每10 min收集流出液10 mL。然后,再向层析柱注入大约5个孔隙体积数(PV)的上述nTiO2悬浮液,用自动部分收集器收集20管流出液。当注入悬浮液后继续注入5 PV的NaNO3背景溶液,收集流出液,待测试。
1.4 钛和磷浓度的分析与测定
钛(Ti)的分析与测定:取2 mL nTiO2悬浮液置于25 mL的烧杯中,向烧杯中加入1-2 mL硫酸-硫酸铵消解液,置于加热板上,在220℃下加热1-1.5 h。待消解完全后,溶液转移至50 mL的容量瓶中定容。从中取5 mL 移入 50 mL 容量瓶中,依次分别加入 8 mL (V盐酸∶V去离子水=5∶1)稀盐酸,2 mL(10 g·L-1)抗坏血酸,10 mL二安替比林甲烷盐酸溶液,定容。用紫外分光光度计(TU-1901,日本岛津)在390 nm波长下测定Ti的浓度。用 1 000 mg·L-1的钛标准储备溶液(基质为 0.15 mol·L-1HNO3)稀释成(浓度梯度为 1-5 mg·L-1)一系列标准溶液,然后得到标准曲线并测得出钛的浓度。
总磷的分析与测定:用钼蓝显色法测定磷浓度[12],把待测液于50 mL的容量瓶中定容,依次分别加入一滴酚酞、一滴 1 mol·L-1NaOH 溶液(摇匀)、一滴 1 mol·L-1硫酸溶液(摇匀至无色),1 mL(100 g·L-1)抗化血酸和2 mL钼酸盐定容,显色20 min后用紫外分光光度计在700 nm波长下测定磷(P)的吸光度。另外,用磷标准储备液稀释成浓度梯度为1-5 mg·L-1的系列标准溶液,相同波长条件下测吸光度,然后获得标准曲线并测定出磷的浓度。
溶解磷的分析与测定:取5 mL nTiO2悬浮液于7 mL高速离心管中,将离心管置于超高速离心机(GL-21M,赛默飞世科技公司)中,在4℃、15 000 r·min-1转速下离心1 h。将上清液过0.22 μm多孔过滤膜,按照上述测试总磷的方法对溶解磷的浓度测试。
1.5 两点动力学模型
选用两点动力学吸附模型(TSKAM),其方程为[13-14]

其中,θ是石英砂柱的孔隙率,C表示溶液中nTiO2颗粒的浓度,ρb表示石英砂的容重,x表示空间垂直坐标轴,D表示水力学弥散系数,ν表示水流速度,S1和S2分别表示滞留位点l和位点2。TSKAM模型的核心是将石英砂表面上利于nTiO2颗粒吸附的位点分为位点1和位点2,滞留在位点2上的nTiO2颗粒受对流-弥散控制,质量守恒方程为为一阶动力学吸附、解吸方程

k2和k2d分别为位点2上的吸附、解吸速率,nTiO2颗粒在位点2上的吸附属于可逆吸附。位点1上的质量守恒方程为

k1为胶体在位点1上的吸附速率。位点1上的吸附是不可逆吸附。ψx是与填充柱深度有关的函数[13]

其中dc是石英砂的平均粒径,x0是坐标轴上的距离,在此距离时nTiO2颗粒滞留与柱深度有关,β是经验系数,控制着空间滞留曲线的形状。位点2吸附效率(k2)、解析效率(k2d)以及位点1吸附效率(k1)值越小,表明颗粒在石英砂上的滞留越少、其迁移性越高。通过HYDRUS-1D软件模拟nTiO2颗粒的穿透曲线,从而得到参数 k1、S1、k2及 k2d[15]。
1.6 迁移参数
nTiO2颗粒质量回收率可以通过对其迁移曲线进行面积积分得到

式中,Q 为孔隙流速(mL·min-1),C0和 C 分别为入流液和出流液 TiO2浓度(mg·L-1),t为时间(min),t0为脉冲持续时间(min)。
nTiO2颗粒在石英砂表面吸附的概率称之为吸附效率(α)

式中,L为柱子的长度,θ为填充柱的孔隙率,dc为砂子的直径,η0为理论单一介质接触效率。净床渗透系数

颗粒物沉积速率系数

式中,νp为水流速度,k为时间和距离相关的系数。
nTiO2颗粒的最大迁移距离定义为当99.9%的nTiO2颗粒被截留时候nTiO2颗粒所移动的距离,即为

以上公式的各种迁移参数结果详见表1。

表1 迁移实验中nTiO2颗粒、石英砂柱的物理和计算参数
2 结果与讨论
2.1 不同浓度NaNO3电解质对nTiO2表面Zeta电位的影响
在pH=6.5时,nTiO2在不同浓度NaNO3中其表面Zeta电位的变化如图1(a)所示。在不同电解质浓度下,nTiO2颗粒表面的Zeta电位都是负值,这表明在不同浓度NaNO3下,nTiO2颗粒的表面均为负电荷。随着溶液中NaNO3浓度的不断增加,nTiO2颗粒表面Zeta电位降低(绝对值降低,即负的越来越少)。当NaNO3浓度从0.1 mM增加到5 mM时,相应的Zeta电位从-19 mV变至-6.09 mV。这主要是因为随着溶液中NaNO3的溶度的不断增加,nTiO2颗粒表面的电荷屏蔽效应和静电双电层被压缩[16-17],nTiO2颗粒表面的净负电荷减少。从而导致nTiO2颗粒表面Zeta电位降低,因此,nTiO2颗粒悬浮液的弥散稳定性也降低。
2.2 不同磷浓度对nTiO2表面Zeta电位的影响
如图1(b)所示,在pH=6.5,背景溶液NaNO3为10 mM时,nTiO2颗粒表面的Zeta电位随着P浓度的增加而增大。例如当P浓度为0.1 mM时,其表面Zeta电位为-28.6 mV,而将P浓度增加到5 mM时,它的表面Zeta电位则增加到-32.43 mV。结果表明:由于磷酸盐吸附在nTiO2颗粒的表面,通过表面羧基的去质子化,提高了nTiO2颗粒表面的电荷密度[18]。因此,nTiO2颗粒和吸附P的nTiO2颗粒之间静电排斥作用加强[19],最终导致nTiO2颗粒悬浮液弥散稳定性的提高。

图 1 不同浓度NaNO3溶液(a)以及10 mM NaNO3背景溶液中不同磷浓度(b)对nTiO2颗粒的Zeta电位(pH=6.5)
2.3 水流速度对悬浮于磷酸盐溶液中nTiO2颗粒迁移的影响
探索悬浮液pH为6.5时,磷酸盐浓度为0.1 mM,背景溶液NaNO3浓度为10 mM时,不同水流速度(0.5-2.5 mL·min-1)对nTiO2颗粒和P在石英砂柱中迁移的影响。在这组实验所选用的水流速度均在地下水的水流速度范围内。图2为不同水流速度时,nTiO2颗粒的穿透曲线。随着水流速度的增加,nTiO2颗粒的出流比(C/C0)不断提高。当水流速度从0.5 mL·min-1升至2.5 mL·min-1,nTiO2颗粒的出流比从3.9%增加至38.0%左右,这种规律与不同水流速度对纳米羟基磷灰石在石英砂柱中的迁移规律相似[21]。产生上述现象主要是因为随着石英砂柱中的水流速度的增加,石英砂柱中石英砂表面可以用来被nTiO2颗粒吸附的总位点也随之减少,当水流速度很大时,由于水力学剪切力的作用,石英砂表面用于被nTiO2颗粒吸附的总位点急剧减少,nTiO2颗粒在石英砂柱中的滞留量也不断减少。此外,当水流速度很低时,在孔隙度很小的石英砂柱中,已经很难提供nTiO2颗粒足够穿透石英砂柱的动能,大量的nTiO2颗粒滞留在石英砂柱中[21-23]。

图2 不同水流速度时nTiO2颗粒的穿透曲线
此外,从表1可以看出,当水体流速从0.5 mL·min-1增加到2.5 mL·min-1时,nTiO2颗粒在石英砂表面的吸附效率从3.56×10-4降低到1.06×10-4,nTiO2颗粒在石英砂柱中的滞留量减少,迁移能力不断提高。虽然沉积速率系数从2.705 h-1增加至7.699 h-1,但是由于水流速度增加,水力学剪切力不断提高,在迁移过程中活性碰撞不断提高,nTiO2颗粒在石英砂表面的滞留量不断减少,更多的nTiO2颗粒穿透石英砂柱。另外,nTiO2颗粒的最大迁移距离随着水流速度的提高,也在不断的增加,并且最大迁移距离均大于柱子的高度17.5 cm,这出说明了nTiO2颗粒在此三种不同水流速度下,均能够顺利的穿透石英砂柱,因此,水流速度的提高促进了nTiO2颗粒在石英砂柱中的迁移。至于水流速度对nTiO2颗粒在饱和石英砂柱中的迁移影响,两点动力学模型可以很好的模拟nTiO2颗粒在石英砂柱中的穿透曲线。如表2所示,水流速度为0.5-2.5 mL·min-1,模拟得到R2分别为0.983、0.996和0.990,说明模型拟合度很高。随着水流速度的增加,位点1(k1)、位点2(k2)的吸附效率和位点1上的一阶解吸速率(k1d)均降低,这表明nTiO2颗粒在石英砂表面吸附的更少了,导致其迁移量增加,更有助于其迁移。

表2 不同实验条件下两点动力学吸附模型模拟参数
2.4 水流速度对磷酸盐迁移的影响
如图3,随着水流速率的增加,总P的出流比不断提高。当水流速率为0.5 mL·min-1时,总P的出流比为38.0%;当水流流速增加到1 mL·min-1时,此时总P的出流比为50%,继续增加水流速度到2.5 mL·min-1,总P的出流比增加至67.9%。这主要是因为随着石英砂柱中的水流速度的增加,nTiO2颗粒穿透石英砂柱的能力增加,这样使得吸附在nTiO2颗粒的P也随之迁移出石英砂柱,消解后,测得总P的浓度也随之增加。但是,随着水流速度的增加,溶解态P的出流比没有明显的变化,如图4所示。此时,溶解态P的出流比基本保持在21.5%左右。溶解态P出流比没有明显的变化,主要是因为水流速度的变化不会影响nTiO2颗粒对P的吸附能力,所以不管水流速度如何变化,溶液中的溶解态P的浓度都不会改变。此外,溶解态P是由总的磷酸盐和nTiO2颗粒结合态P相减所得。因此,在改变速度,溶解态P浓度不变的情况下,在水流速率为0.5 mL·min-1时,出流液中溶解态P占主要,只有少部分与nTiO2颗粒结合态P存在。当水流速率提高后,此时出流液中的P大部分均以与nTiO2颗粒结合态P的形式存在,只有少部分以溶解态P的形式存在。

图3 不同水流速度对悬浮于(0.1 mM)磷酸盐溶液中总磷的穿透曲线
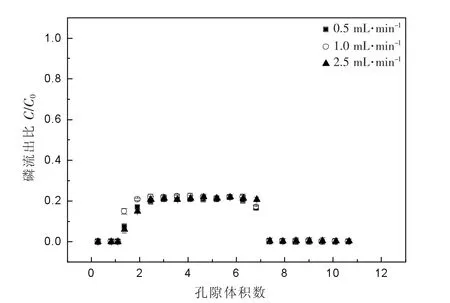
图4 不同水流速度对悬浮于(0.1 mM)磷酸盐溶液中溶解态磷的穿透曲线
3 结语
(1)在pH=6.5时,随着电解质NaNO3浓度的增加,nTiO2颗粒表面Zeta负电位逐渐减少;(2)nTiO2表面负电荷随着P浓度的增加而增加,其弥散稳定性也随之提高;(3)高水流流速促进nTiO2的迁移性,并且可溶性磷酸盐的迁移量也不断增加;(4)两点动力学吸附模型能够很好地模拟纳米材料在石英砂柱中的迁移穿透曲线。模型结果表明,随着流速的增加,导致位点2吸附效率(k2)和解析效率(k2d)以及位点1吸附效率(k1)的减小,nTiO2颗粒石英砂上的吸附效率降低,从而更多的nTiO2颗粒穿透石英砂柱。
[1]FANG J,SHAN X Q,WEN B,et al.Stability of titania nanoparticles in soil suspensions and transport in saturated homogeneous soil columns[J].Environmental Pollution,2009,157(4):1101-1109.
[2]HIGARASHI M M,JARDIM W F.Remediation of pesticide contaminated soil using TiO2,mediated by solar light[J].Catalysis Today,2002,76(2-4):201-207.
[3]NAGAVENI K,SIVALINGAM G,HEGDE M S,et al.Photocatalytic degradation of organic compounds over combustion-synthesized nano-TiO2[J].Environmental Science&Technology,2004,38(5):1600-1604.
[4]QUAN X,ZHAO X,CHEN S,et al.Enhancement of p,p’-DDT photodegradation on soil surfaces using TiO2induced by UV-light[J].Chemosphere 2005,60(2):266-273.
[5]AND T A,MADRAS G.Photocatalytic degradation of rhodamine dyes with nano-TiO2[J].Industrial&Engineering Chemistry Research,2007,46(1):1-7.
[6]WEI J,HAMID M,BAOSHAN X.Bacterial toxicity comparison between nano-and micro-scaled oxide particles[J].Environmental Pollution,2009,157(5):1619-1625.
[7]HUND-RINKE K,SIMON M.Ecotoxic effect of photocatalytic active nanoparticles (TiO2) on algae and daphnids[J].Environmental Science&Pollution Research,2006,13(4):225-232.
[8]SALEH N,KIM H,PHENRAT T,et al.Ionic strength and composition affect the mobility of surface-modified FeO nanoparticles in water-saturated sand columns[J].Environmental Science&Technology,2008,42(9):3349-3355.
[9]FRENCH R A,JACOBOSON A R,BOJEONG K,et al.Influence of ionic strength,pH,and cation valence on aggregation kinetics of titanium dioxide nanoparticles[J].Environmental Science&Technology,2009,43(5):1354-1359.
[10]HEALY K E,DUCHEYNE P.Hydration and preferential molecular adsorption on titanium in vitro[J].Biomaterials,1992,13(8):553-561.
[11]KAUSHIK R D,GUPTA V K,SINGH J P.Distribution of zinc,cadmium,and copper forms in soils as influenced by phosphorus application[J].Arid Soil Research&Rehabilitation,2009,7(2):163-171.
[12]CHEN J Y,GAO F X,SUN X Y.Determination of phosphorus content in alcoholic by molybdenum blue extraction photometric method[J].Chemical Engineer,2005,115(4):29-30.
[13]SCHIJYEN J F,SIMUNEK J.Kinetic modeling of virus transport at the field scale[J].J Contam Hydrol,2002,55(1-2):113-135.
[14]BRADFORD S A,SIMUNEK J,BETTAHAR M,et al.Modeling colloid attachment,straining,and exclusion in saturated porous media[J].Envi ronmental Science&Technology,2003,37(10):2242-2250.
[15]MARQUARDT D W.An algorithm for least-squares estimation of nonlinear parameters[J].Journal of the Society for Industrial&Applied Mathematics,2006,11(2):431-441.
[16]ELIMELECH M,GREGORY J,JIA X,et al.Particle deposition and aggregation:Measurement modeling and simulation[M].Woburn:Buaerworth-Heinemann,1995.
[17]HUNTER R J.Foundations of colloid science[M].New York:Oxford University Press,1987.
[18]SOLOVITCH N,LABILLE J,ROSE J,et al.Concurrent aggregation and deposition of TiO2nanoparticles in a sandy porous media[J].Environmental Science&Technology,2010,44(13):4897-4902.
[19]PELLEY A J,TUFENKJI N.Effect of particle size and natural organic matter on the migration of nano-and microscale latex particles in saturated porous media[J].Journal of Colloid&Interface Science,2008,321(1):74-83.
[20]WANG D,BRADFORD S A,PARADELO M,et al.Facilitated transport of copper with hydroxyapatite nanoparticles in saturated sand[J].Soil Science Society of America Journal,2012,76(2):375-388.
[21]GARGIULO G,BRADFORD S A,SIMUNEK J,et al.Transport and deposition of metabolically active and stationary phase deinococcus radiodurans in unsaturated porous media[J].Environmental Science&Technology,2007,41(4):1265-1271.
[22]GARGIULO G,BRADFORD S A,SIMUNEK J,et al.Bacteria transport and deposition under unsaturated flow conditions:the role of water content and bacteria surface hydrophobicity[J].Vadose Zone Journal,2008,7(2):406-419.
[23]BRADFORD S A,TORKZABAN S,WIEGMANN A.Pore-scale simulations to determine the applied hydrodynamic torque and colloid immobilization[J].Vadose Zone Journal,2010,10(1):252-261.
