儿童Prader-Willi综合征并发代谢综合征1例并文献复习
2018-03-15方海宁
方海宁,于 飞
(湖北省妇幼保健院儿童内分泌遗传代谢科,湖北 武汉 430070)
Prader-Willi综合征(Prader-Willi syndrome,PWS)是由Prader等于1956年首先报道的一种多系统异常综合征,又称为肌张力低下-智能障碍-性腺发育滞后-肥胖综合征。虽然Holm等在1993年提出了PWS的临床诊断标准,但源于中国广大的地域差别及基因检测技术推广的局限性,仍有相当比例的患儿在生后数年,甚至到青少年期、成人期才被确诊。本文对1例确诊为PWS合并多种代谢紊乱9岁患儿的临床资料及基因检测结果进行分析,并对近年来国内外的有关文献进行复习。
1病例资料
1.1病史情况
患儿男,9岁1个月,因“体重迅速增长2年”于2015年10月收入湖北省妇幼保健院。患儿为G5P2(母亲选择性流产3次,孕期无保胎)足月顺产,出生体重3.6kg,出生时严重窒息,在当地医院新生儿科抢救15天出院;生后混合喂养,有喂养困难,有肌张力低下,曾在当地医院反复行脑瘫康复训练。患儿5个月抬头,6个月翻身,2岁坐,3岁走,语言、智力发育落后,生后至近2年前身高、体重与同龄、同性别儿相比为中上等。患儿近2年食量明显增加,体重迅速增长,约20kg/年,伴生长加速,约10cm/年;有头晕、头痛,夜间不能平卧,有呼吸费力,需半卧位及辅助翻身,并于半年前在当地医院查血压升高,最高160/110mmHg,间断口服氨氯地平、尼群地平降压治疗,血压未能降至正常。
既往于2009年行隐睾手术,于5岁左右有不明原因发热持续1个月,有情绪激动、易激惹及攻击行为。家族中外祖父、祖父有高血压病,均中年起病。入院体检:体温36.5℃,呼吸30次/分,脉搏100次/分,血压120/88mmHg,身高134.5cm,体重97kg,体质量指数(BMI)53.6kg/m2,臀围138.5cm,腹围125cm,头发浅棕色,眼裂偏小,上翘,虹膜浅黄色,腭弓稍高,双乳B1期,双肺呼吸音稍粗,无啰音,心音有力,腹部、腰间、下肢可见紫纹,双腹股沟区可见手术疤痕,右侧睾丸容积2mL,左阴囊至左腹股沟区可及15cm×13cm×8cm包块,质软,可还纳,左侧睾丸未触及,阴茎埋藏,双侧通贯手,左足内翻,双下肢外旋,扁平足,生理反射可引出,病理征阴性,见图1。
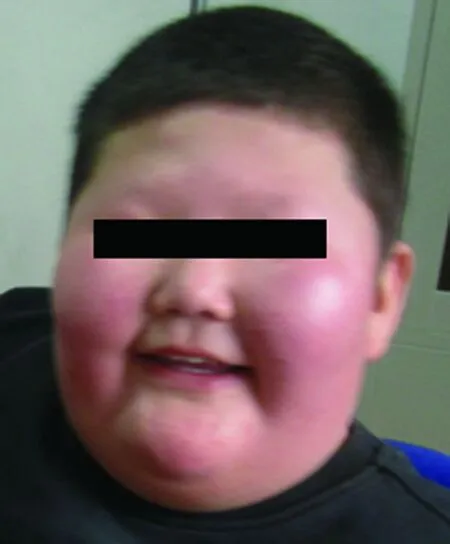
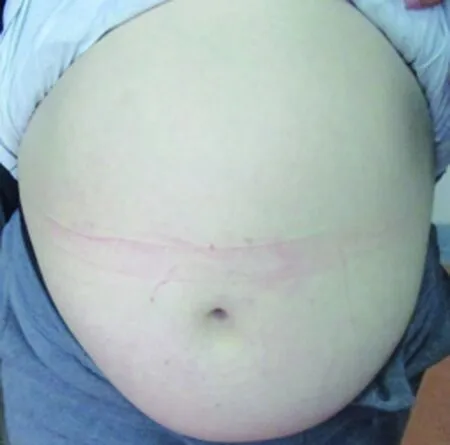
图1 本例PWS患儿临床表型特征
Fig.1 Clinical phenotypic characteristic of the case with PWS
1.2辅助检查
1.2.1一般检查
肝功能正常。肾功能:尿酸424μmol/L,偏高。血脂:低密度脂蛋白胆固醇2.73mmol/L,升高;高密度脂蛋白胆固醇0.93mmol/L,下降;糖化血红蛋白6.3%。甲状腺功能:正常范围。立位肾素-血管紧张素-醛固酮系统(RAAS):正常范围;卧位RAAS:血管紧张素Ⅱ7 076pg/mL,升高,皮质醇节律正常。性激素全套:睾酮0.068ng/mL,卵泡刺激素3.52mIU/mL,黄体生成素0.5mIU/mL,雌激素11.15pg/mL,泌乳素10.8ng/mL,尿微量白蛋白81mg/L,升高。口服葡萄糖耐量试验提示患儿正常糖耐量,胰岛素正常范围。戈拉瑞林刺激试验提示患儿处于青春期前水平。精氨酸及可乐定复合生长激素激发试验提示患儿生长激素峰值0.225ng/mL。
1.2.2特殊检查
垂体MRI平扫+增强:垂体体积偏小。肝胆脾彩超:患儿皮下脂肪增厚,透声差,显示不清。肝胆脾胰双肾MRI:普遍重度脂肪肝。肺部CT:未见异常。心脏彩超:左心扩大。主动脉、肾动脉、冠状动脉CTA:未见明显血管狭窄、畸形,室间隔增厚,纵膈、心包周围脂肪沉积。肾上腺CT增强:未见异常。会阴部B超:类似阴囊突出物内未见正常睾丸。下腹部MRI:左腹股沟斜疝,睾丸体积偏小,明显向右推移。骨龄:10~11岁。肺功能:中度限制性通气障碍。睡眠呼吸监测:阻塞性睡眠呼吸暂停低通气综合征(重度)。外周血染色体核型分析:46,XY。染色体微阵列分析(CytoScan HD基因芯片):拷贝数变异(copy number variant,CNV)类型缺失,染色体与基因组位置arr[hg19]15q11.2q13.1(22 770 421~28 823 722)×1,CNV大小6.1Mb,见图2。
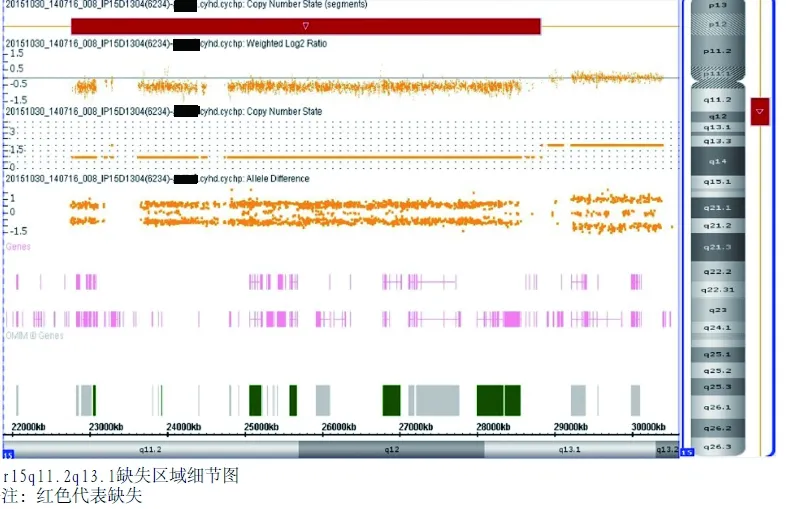
图2 染色体微阵列分析
Fig.2 Chromosomal microarray analysis
1.3诊断
Prader-Willi综合征、代谢综合征(metabolic syndrome,MS)(高血压病、高血压心脏病、重度脂肪肝、睡眠呼吸暂停综合征)、埋藏阴茎、左腹股沟疝。
1.4治疗
给予①控制血压:卡托普利(开博通)12.5mg,2次/日,苯磺酸氨氯地平(络活喜)5mg,1次/日,口服;②改善胰岛素抵抗:二甲双胍肠溶片(格华止)0.85g,2次/日,口服;③抗血小板聚集:阿司匹林片150mg,1次/日,口服;④抑制食欲、控制体重、改善胰岛素抵抗:艾塞那肽针(百泌达)5μg,早晚餐前30分钟皮下注射。患儿血压控制110/70mmHg左右,夜间睡眠改善,体重停止增加出院。
2讨论
2.1 Prader-Willi综合征是一类基因组印迹性疾病
人类个体从父母获得两套完整的染色体,一套来源于父亲,另一套来源于母亲,而多数常染色体表达父母双方的等位基因,印迹基因仅表达等位基因中的一个。自从20世纪80年代研究基因组印记及其相关疾病以来,首次报道的基因组印迹性疾病就是肌张力低下-智能障碍-性腺发育滞后-肥胖综合征[1]。人类父源15号染色体q11-13区域异常是导致疾病发生的原因[2]。Prader-Willi综合征分子缺陷类型包括[3]:①染色体15q11-q13父源基因缺失(deletion)约占70%;②母源单亲二体(uniparental disomy,UPD)指含有2条母源染色体,约占25%;③父源等位基因印迹缺陷(imprinting defects)主要是由印迹基因突变所致,只占1%~3%;④染色体平衡易位及其它罕见原因小于1%。由于DNA分子CpG二核苷酸是调节基因功能的关键原件,而基因组印迹基因主要与DNA分子CpG二核苷酸中的胞嘧啶甲基化有关。鉴于这种特性,近年来建立和完善了甲基化聚合酶链反应(methylation-specific polymearse chain reaction,MS-PCR)技术检测进行基因诊断的方法。此前被用于基因检测的方法还包括高分辨染色体分析和荧光原位杂交(fluorescence in situ hybridization,FISH)法,其中MS-PCR能检测出缺失、单亲二体和印迹缺陷三种分子缺陷类型,被认为是高效、快速,特异性、敏感性均佳的检测方法,其基本原理是基于15号染色体q11-13区域SNRPN基因序列设计的,故而对15号染色体的微缺失特异性高,但Geets等[4]研究提出6q14.1-6q16.3的缺失也可能导致类Prader-Willi综合征样的症状,而CMA技术可以通过全基因组水平的扫描检测染色体不平衡的CNV,故而可以覆盖包括15号染色体在内的所有引起肥胖的类Prader-Willi综合征的染色体拷贝数变异,尤其是对于检测染色体组微小缺失、重复等不平衡性重排具有突出优势。根据芯片设计与检测原理的不同,CMA技术可分为两大类:基于微阵列的比较基因组杂交(array based comparative genomic hybridization,aCGH)技术和单核苷酸多态性微阵列(single nucleotide polymorphism array,SNP array)技术。Morandi等[5]报道了1例MS-PCR检测正常,SNP array技术检测提示15号染色体母源单亲二体的病例,故MS-PCR有时尚需与SNP array技术相互印证。但CMV技术也存在固有的局限性,即无法可靠地检出低水平的嵌合体及平衡性染色体重排和大多数的基因内点突变[6]。因此,在对PWS的检测策略选择上,必要时需多管齐下。本例患儿即是采用CMA技术明确了染色体缺失的位置及大小,结合临床症状,可以明确诊断PWS。
2.2 Prader-Willi综合征的临床表现各异
PWS的临床特征在不同年龄段表现不同,甚至基因型不同,临床特点亦可有较大差别,例如UPD患者可能没有典型的面部特征。PWS主要表现为两个阶段:新生儿期的肌张力低下、吸吮无力、喂养困难、少哭少动,童年期及以后出现的过度饮食、肥胖、矮小、性腺发育障碍等,其为与生长发育及内分泌有关的临床综合征[7]。本资料中患儿临床症状符合Holm等在1993年提出的PWS临床诊断标准中所有的8条主要标准,5条次要标准,评分大于8分,结合患儿染色体微阵列分析结果,患儿15q11.2q13.1发生了6.1Mb的缺失,患儿PWS诊断确立。尽管该患儿在新生儿期症状即有显著显现,但鉴于地域特点及当时对该疾病认识的局限性,未能实现早期诊断,患儿因肥胖、高血压再次在当地医院,亦未能明确诊断该疾病,故儿科医生早期识别该疾病的临床特征至关重要。Lu等[8]研究了31名中国PWS患者,所有患者均有新生儿期肌张力低下及喂养困难,但仅12.9%的患者有身材矮小,54.8%有特殊面容,35.5%有皮肤损伤,提示中国患儿身材矮小、特征性面容、自我皮肤损伤和母源性UPD型的发生率比西方PWS患者的发生率低。尽管该患儿生长激素激发试验显示生长激素水平绝对缺乏,但骨龄超过实际年龄接近2岁,因骨龄提前所增加的身高弥补了生长激素缺乏所致的生长落后,故而未出现典型的身材矮小,使得患儿身高处于同龄同性别儿童中位数水平,骨龄的提前考虑为肥胖所引起的脂肪组织增多,使脂肪组织中芳香化酶催化雄激素转化为雌激素增加,而雌激素则会促进钙磷在骨骼的沉积,促进骨骼的生长和骨骺的闭合。因此,虽然Holm等提出的临床诊断标准中主要标准包含生长落后或矮小,但身高的绝对值决不能作为PWS的排除条件,需结合患儿发育时期、骨龄、肥胖程度等情况综合考虑。
大多数研究者把PWS肥胖的原因归结为饱腹感异常导致的摄食量增加[9]。有学者发现在PWS的儿童和成人患者中,一种名为ghrelin的饥饿激素水平明显升高[10],并且强有力地增加了人的食欲,参与胃口、体重和生长激素分泌的调节[11]。Chen等[12]在一项针对伴有肥胖的PWS白种人的基因研究中提出有3个基因与体脂增加有关,这3个基因位于15q11.2,在两个已知的PWS印记基因NDN和C15orf2之间,该学者指出PWS印迹基因本身与肥胖并无关联,但位于印迹基因之间的一种新的基因名为“PWRN1”(Prader-Willi区非蛋白编码RNA1)与肥胖有关。但也有对非肥胖体型PWS的解释,Hirsch等[13]的研究指出PWS患者血浆唾液中的irisin含量明显升高,而irisin能促进白色脂肪组织向棕色脂肪组织的转化,从而激发能量代谢,降低体重增加的风险。尽管目前对于PWS导致肥胖的发病机制进行了深入探讨,但导致PWS肥胖与否的原因至今尚未明确。
本资料中患儿5岁时有出现不明原因发热持续1个月。在国内外文献中也均有报道PWS患儿出现体温调节紊乱的症状[14]。结合患儿超量摄食、促性腺激素水平低下,PWS患儿的多种临床症状符合下丘脑功能异常所致的一系列后果,因为下丘脑正是体温调节中枢、摄食中枢、发育中枢等关键部位所在。
2.3 Prader-Willi 综合征与代谢综合征
代谢综合征(MS)是一组与中心性肥胖、糖尿病、心脑血管疾病密切相关的代谢异常或危险因素的综合征,主要表现为中心性肥胖、高血压、糖脂代谢紊乱、胰岛素抵抗、微量蛋白尿、高尿酸血症、血液高凝状态(纤维蛋白原增加)以及非酒精性脂肪肝病等。2007年国际糖尿病联合会(International Diabetes Federation,IDF)提出了全球统一的儿童代谢综合征诊断标准,并将中心性肥胖作为代谢综合征的必备条件,以高血压、高血糖、高TG、低HDL-C为选择组分,同时进行了适宜年龄范围的划分。2012年中国医学会儿科分会提出了中国儿童青少年代谢综合征诊断标准。虽然本资料中患儿就诊时9岁1个月,但至今已到达10周岁,并完全符合肥胖症、高血压、血脂异常的标准,其尿微量白蛋白及尿酸亦偏高,合并重度脂肪肝及重度睡眠呼吸暂停综合征,有多种代谢紊乱及高危因素,有进展为心血管疾病、脑血管疾病甚至猝死的高风险。患儿的高血压经影像学及实验室检查的证实可排除继发因素,考虑为原发性高血压。肥胖本身就是原发性高血压的危险因素,血压与体重及BMI呈正相关[15],肥胖同样会增加糖耐量异常、血脂异常等一些代谢并发症发生的危险[16],而且是导致胰岛素抵抗的主要原因[17]。有国外学者在对109例PWS儿童的研究中指出:PWS总的代谢综合征的发病率为7.3%,但是非肥胖的PWS没有出现代谢综合征,而肥胖的PWS中代谢综合征的发病率可达16%,提示肥胖状态是PWS发生代谢综合征的中心环节[18]。值得一提的是,本资料中患儿的腹部影像学提示的重度脂肪肝、心包周围脂肪沉积,证实了其肥胖类型为腹型肥胖,即内脏脂肪的分布以腹内脏器为主,内脏脂肪堆积导致代谢紊乱和心血管疾病发生的作用独立于整体脂肪含量,显示了腹型肥胖在代谢综合征中的重要地位[19]。尽管该患儿目前的胰岛素水平并未出现胰岛素抵抗的情况,但可以预见其发展为2型糖尿病的风险明显升高。也有学者提示PWS患者显示出更好的代谢指标[20](更高的胰岛素敏感性,更低的腰围、空腹血糖水平、胰岛素抵抗指数、胆固醇、转氨酶水平和躯干脂肪/脂肪的质量比)。因此,在看待儿童PWS与代谢综合征的关系时,应充分考虑到儿童及青少年PWS肥胖出现的年龄波动范围会较大,并有成年后发病的可能。从流行病学上看,肥胖仍是PWS患者最主要的死亡原因,且肥胖与睡眠呼吸暂停综合征互为因果,肥胖相关的心脏和呼吸系统疾病是15岁以后患者的常见死因[21]。因此,对于PWS患儿在早期诊断的同时需首先重视监测控制体重,避免向后续的并发症进展,才能最大可能延长预期寿命至接近正常[22]。
2.4结论
PWS的发病率从1/500到1/15 000不等,常在新生儿期被漏诊、误诊,故新生儿期出现的的肌张力低下、喂养困难和生殖器官发育不全的患儿,临床上应怀疑为PWS病,必要时尽早行基因分析。在肥胖的儿童、青少年中,PWS也是一个潜在的病因,应关注肥胖儿童的摄食情况、认知和语言等神经发育情况以及性腺发育状况,从中筛查出可疑的PWS,因为在超重或肥胖发生前的体重干预能极大的改善预后,延长寿命。尽管PWS可能有特异的外貌及典型的症状,但不能忽视种族的差异与表型的多样性。对PWS确诊的金标准有赖于基因分析发现15号染色体q11-13区域的异常,现有的MS-PCR技术能确诊绝大部分患者,但需考虑到每项检测均有其技术上的缺陷性,对于临床症状高度符合但MS-PCR阴性的患者,不应马上排除,可考虑其他基因检测手段的综合应用,同时应关注类Prader-Willi综合征的存在,CMV技术有助于发现类Prader-Willi综合征。PWS有发展为重度肥胖,并由此造成代谢综合征的危险,从而严重影响患儿的生存质量及预期寿命,故应加强对PWS患儿发生代谢综合征的危险因素早期识别及早期干预,特别是肥胖这一诱导患儿发展为代谢综合征的中心问题。
[1]廖二元.内分泌代谢病学[M].3版.北京:人民卫生出版社,2012:293.
[2]Bajrami E, Spiroski M.Genomic imprinting[J].Open Access Maced J Med Sci,2016,4(1):181-184.
[3]高力敏,石岩.Prader-Willi综合征分子遗传学诊断[J].中国医疗前沿,2011,6(2):73-74.
[4]Geets E, Zegers D, Beckers S,etal. Copy number variation (CNV) analysis and mutation analysis of the 6q14.1-6q16.3 genes SIM1 and MRAP2 in Prader Willi like patients[J].Mol Genet Metab,2016,117(3):383-388.
[5]Morandi A, Bonnefond A, Lobbens S,etal. A girl with incomplete Prader-Willi syndrome and negative MS-PCR, found to have mosaic maternal UPD-15 at SNP array[J].Am J Med Genet A,2015,167A(11):2720-2726.
[6]染色体微阵列分析技术在产前诊断中的应用协作组.染色体微阵列分析技术在产前诊断中的应用专家共识[J].中华妇产科杂志,2014,49(8):570-572.
[7]Ramsden S C, Clayton-Smith J, Birch R,etal. Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes[J].BMC Med Genet,2010,11:70.
[8]Lu W, Qi Y, Cui B,etal. Clinical and genetic features of Prader-Willi syndrome in China[J]. Eur J Pediat,2014,173(1):81-86.
[9]Naik S, Thomas N S, Davies J H,etal.Novel tandem duplication in exon 1 of the SNURF/SNRPN gene in a child with transient excessive eating behaviour and weight gain[J]. Mol Syndromol,2012,2(2):76-80.
[10]Kuppens R J, Diène G, Bakker N E,etal.Elevated ratio of acylated to unacylated ghrelin in children and young adults with Prader-Willi syndrome[J].Endocrine,2015,50(3):633-642.
[11]Mihalache L, Gherasim A, Nitǎ O,etal.Effects of ghrelin in energy balance and body weight homeostasis[J].Hormones (Athens),2016,15(2):186-196.
[12]Chen Y, Liu Y J, Pei Y F,etal. Copy number variations at the Prader-Willi syndrome region on chromosome 15 and associations with obesity in whites[J].Obesity(Silver Spring),2011,19(6):1229-1234.
[13]Hirsch H J, Gross I, Pollak Y,etal.Irisin and the metabolic phenotype of adults with Prader-Willi syndrome[J].PLoS One,2015,10(9):e0136864.
[14]冯英,肖农,陈玉霞.Prader-Willi综合征体温不稳定1例报告[J].临床儿科杂志,2015,33(4):361-363.
[15]Ma J, Wang Z, Dong B,etal. Body fat and blood pressure: comparison of blood pressure measurements in Chinese children with different body fat levels [J].Br J Nutr,2012,108(9):1672-1677.
[16]Kong A P, Chow C C.Medical consequences of childhood obesity:a Hong Kong perspective[J].Res Sports Med,2010,18(1):16-25.
[17]Jung U J, Choi M S. Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease[J].Int J Mol Sci,2014,15(4):6184-6223.
[18]Brambilla P, Crinò A, Bedogni G,etal.Metabolic syndrome in children with Prader-Willi syndrome:the effect of obesity[J].Nutr Metb Cardiovasc Dis,2011,21(4):269-276.
[19]Kim J, Lee S H. Therapeutic results and safety of postoperative radiotherapy for keloid after repeated Cesarean section in immediate postpartum period[J]. Radiat Oncol J,2012,30(2):49-52.
[20]Fintini D, Inzaghi E, Colajacomo M,etal. Non-Alcoholic Fatty Liver Disease (NAFLD) in children and adolescents with Prader-Willi Syndrome(PWS)[J].Pediatr Obes,2016,11(3):235-238.
[21]Senda M, Ogawa S, Nako K,etal. The glucagon-like peptide-1 analog liraglutide suppresses ghrelin and controls diabetes in a patient with Prader-Willi syndrome[J].Endocr J,2012,59(10):889-894.
[22]Jin D K.Systematic review of the clinical and genetic aspects of Prader-Willi syndrome[J].Korean J Pediatr,2011,54(2):55-63.
