高效液相色谱-串联质谱法快速测定水产品中19种喹诺酮类药物残留
2018-03-06梁晶晶徐潇颖丁宇琦陈万勤罗金文
梁晶晶,徐潇颖,丁宇琦,陈万勤,刘 柱,罗金文
(浙江省食品药品检验研究院,浙江 杭州 310052)
喹诺酮类药物是一种具有4-喹诺酮共同基本母核的人工合成抗菌药,可通过直接作用于细菌的核,抑制其DNA旋转酶,破坏其代谢和繁殖,从而迅速杀灭细菌,具有抗菌谱广、抗菌力强等优点,被广泛应用于动物和人类的多种感染性疾病的预防和治疗[1]。但该类药物对消化系统、中枢神经系统、肌肉、骨骼等有一定毒性且易产生耐药性[2-3],如对动物长期使用,会造成动物性食品中喹诺酮药物残留,同时对食用者的健康造成危害。近年来,部分水产品养殖户为提高产量,常在养殖过程中乱用、滥用此类药物,以致水产品安全事件频发。为确保消费者的安全,我国及欧盟等组织均对水产品中喹诺酮类药物的使用情况及残留限量做了严格规定。如我国规定牛、鸡、猪、羊、兔等动物的肌肉、脂肪、肝、肾中达氟沙星、二氟沙星、 恩诺沙星(环丙沙星与恩诺沙星之和)、沙拉沙星等喹诺酮类兽药的最高残留限量为0.01~1.9 mg/kg[4]。
喹诺酮类药物残留问题已越来越引起人们的重视,其检测方法主要有微生物法[5]、酶联免疫法[6-7]、液相色谱法[8-9]、液相色谱-串联质谱法等[10-12]。但微生物法的检测限过高,特异性不强;酶联免疫法属于半定量筛查方法,易产生假阳性现象;液相色谱法灵敏度低,抗干扰能力弱,定性效果差,且不适合多残留同时分析。近年来,液相色谱-串联四极杆质谱联用(LC-MS/MS)技术由于兼具液相色谱的分离能力和串联质谱的高灵敏度、高专属性特点,正逐渐成为兽药残留分析的有效手段。但由于水产品基质复杂,通常含有较多的蛋白质、脂肪和磷脂,在样品预处理过程中,采用沉淀和离心去除蛋白的同时,脂肪和磷脂也被共提取出。现有的前处理方法通常采用正己烷脱脂、SPE固相萃取柱、离子交换柱或用其他反向吸附剂(如硅胶、C18等)从组织提取物中去除脂肪,以消除样品基质的干扰。但这些前处理方法普遍存在步骤繁琐、测定时间长等缺点,且无法去除磷脂。
PRiME HLB是一种最新型的固相萃取柱,是基于“N-乙烯吡咯烷酮二乙烯基苯聚合物”填料的反相复合填料,对动物源性食品中的蛋白质、脂肪和磷脂等物质具有特异性的吸附能力,且无需活化与平衡,样品经有机溶剂提取后,可直接上样,操作极其简便,不会出现堵柱情况。而目前常用的SPE净化方式(HLB、WAX等)均需经活化、平衡、上样、淋洗和洗脱等多个步骤,操作繁琐,费时费力。PRiME HLB采用最简单的过滤式净化方式,可将净化速度提高40%,只需3个步骤即可完成整个样品制备过程,操作简单、省时快速,净化后可以去除动物源性食品中蛋白、脂肪和磷脂等95%以上的基质干扰物,确保数据的稳定性和可靠性。目前国内外仅有少量将此技术应用于水产品中磺胺类和青霉素类药物残留检测的报道[13-14],但未见其用于水产品中喹诺酮类药物残留检测的相关报道。本研究以明虾和鲈鱼为研究对象,采用PRiME HLB样品净化技术,以超高效液相色谱-串联四极杆质谱(UPLC-MS/MS)为分析手段,同时测定水产品中19种喹诺酮类药物的残留量。该方法简便快速,易于操作,为水产品中喹诺酮类药物残留的测定提供了新途径。
1 实验部分
1.1 仪器与试剂
XevoTMTQ-S液相色谱-串联质谱仪(美国Waters公司);Milli-Q超纯水器(美国Millipore公司);氮气吹干仪(Biotage公司);涡旋混合器(上海琪特公司);Multiduge X1台式离心机(美国Thermo公司);Ks 4000 Ic低温摇床(德国IKA公司);超声波清洗器(昆山禾创超声仪器有限公司)。
甲醇、乙腈(色谱纯,德国Merck公司);甲酸(色谱纯,美国Fluka公司);乙酸铵(质谱纯,美国Sigma公司);Oasis PRiME HLB固相萃取柱(200 mg,6 mL,美国Waters公司)。
实验所用19种兽药的标准物质均购自Dr.Ehrenstorfer GmbH,喹诺酮内标混合液购自北京振翔科技有限公司。
1.2 标准溶液配制
1.2.1对照品储备液分别精密称取19种喹诺酮类药物10 mg(精确至0.01 mg),用乙腈溶解并定容至20 mL,配成500 mg/L的标准储备液,于-18 ℃下存放。
1.2.2混合对照品中间液精密移取0.2 mL各对照品储备液至10 mL容量瓶中,用乙腈定容,精密移取1.0 mL至50 mL容量瓶中,用乙腈定容,即得200 μg/L的混合对照品中间液,于4 ℃下保存。
1.2.3同位素内标储备液直接购买10 mg/L的混合同位素内标储备液(诺氟沙星-D5、萘啶酸-D5、培氟沙星-D5、恩诺沙星-D5、双氟沙星-D3)。
1.2.4混合同位素内标中间液精密移取0.1 mL混合同位素内标储备液,用乙腈定容至5.0 mL,即得200 μg/L的混合同位素内标中间液,于4 ℃下避光保存。
1.2.5混合标准曲线的制备精密移取混合对照品中间溶液和混合同位素内标中间液适量(标准工作溶液中内标物浓度应与样品待测液中一致),用空白样品基质溶液配成0.2~20 μg/L的系列混合标准工作溶液。
1.3 样品前处理方法
准确称取样品2.0 g于50 mL离心管中,精确加入同位素内标混合液0.2 mL。加入9.8 mL 80%乙腈水溶液(含0.1%甲酸),振荡30 min,再以20 000 r/min离心5 min。取2 mL上清液加至PRiME HLB 固相萃取柱,保持1滴/s的流速,待过完后加入2 mL提取液冲洗小柱,收集所有流出液于45 ℃下氮吹至小于0.7 mL,残留液用纯水定容至1.00 mL,过0.22 μm有机滤膜,待上机测试。
空白样品基质溶液的制备:取不含喹诺酮类药物的空白样品,除不加内标工作液外,同上处理,制得空白样品基质溶液。
1.4 色谱与质谱条件
1.4.1色谱条件色谱柱:Waters ACQUITY UPLC BEH C18柱(1.7 μm,2.1 mm×100 mm);流速:0.3 mL/min;柱温:35 ℃;进样量:2 μL;流动相为5 mmol/L乙酸铵水溶液(含0.1%甲酸)(A)和甲醇(B)。梯度洗脱程序:0~0.25 min,2%B;0.25~11.25 min,2%~100%B;11.25~13.00 min,100%B;13.00~13.10 min,100%~2%B;13.10~17.00 min,2%B。
1.4.2质谱条件电喷雾离子源(ESI);正离子扫描模式;多反应监测(MRM)模式;源电压3.5 kV;离子源温度150 ℃;脱溶剂气温度500 ℃,脱溶剂气流量800 L/h;19种待测化合物的质谱参数见表1。
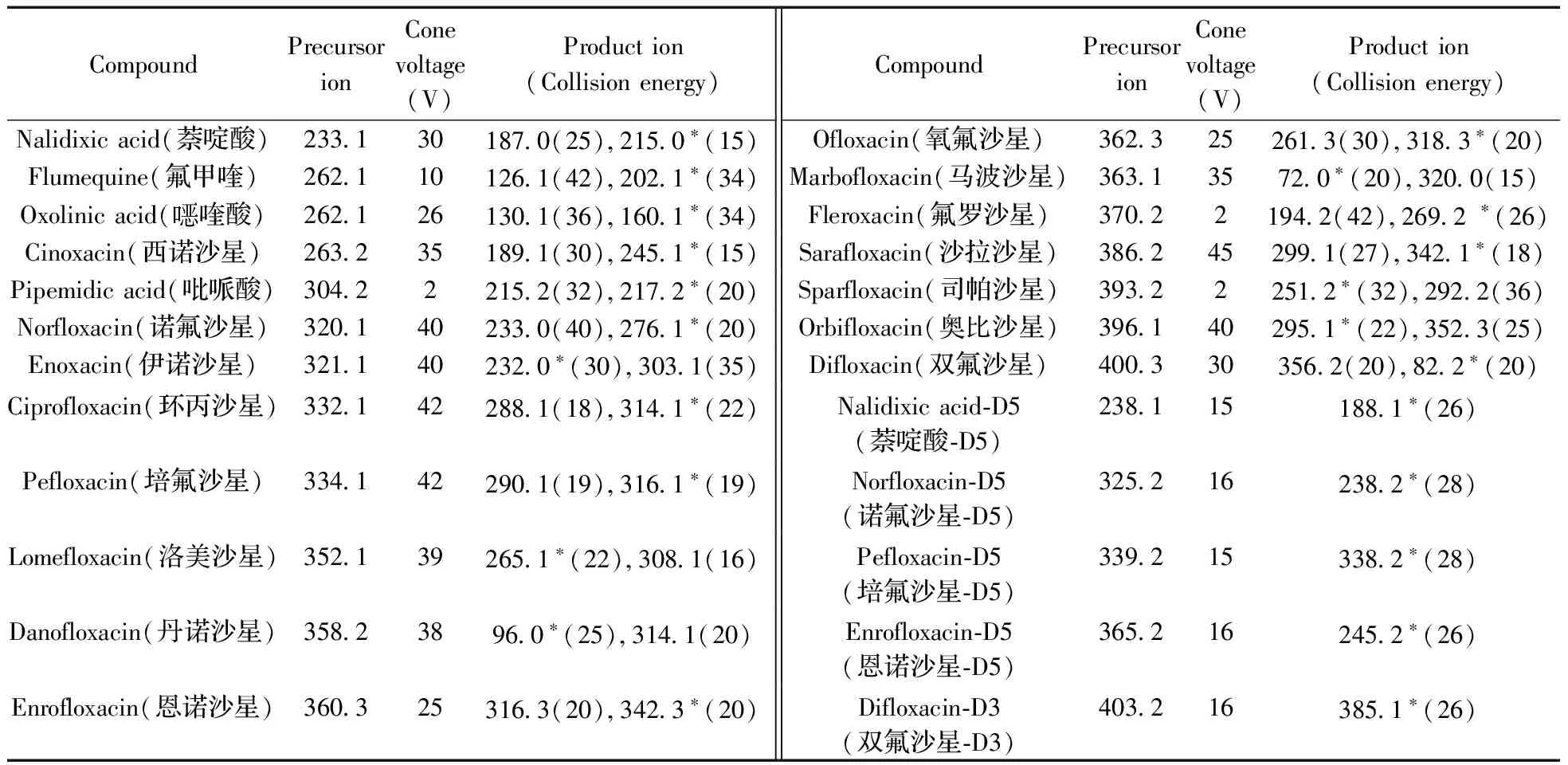
表1 MRM监测模式下19种药物及内标物的质谱条件Table 1 Mass conditions of 19 drugs and internal standard under MRM mode
*quantitation ion
2 结果与讨论
2.1 色谱与质谱条件的优化
选择柱效高、稳定性好、保留能力强的Waters ACQUITY UPLC BEH C18柱(1.7 μm,2.1 mm×100 mm),比较了乙腈-水和甲醇-水两种流动相对19种待测物的分离效果。结果表明,以甲醇-水作为流动相时,分离度和灵敏度明显优于乙腈-水流动相。为进一步提高待测化合物的离子化效率,增加灵敏度,结合相关报道[15],在流动相中添加对正离子有增强作用的甲酸和乙酸铵,通过比较不同甲酸浓度和乙酸铵浓度对灵敏度及峰形的影响,最终确定流动相为甲醇-5 mmol/L乙酸铵水溶液(含0.1%甲酸);再优化流动相比例,最终选择线性梯度洗脱。 上述优化条件下目标物分离度较好、保留时间适中。
通过一级质谱扫描分析0.5 mg/L的19种喹诺酮标准溶液和同位素内标液,得到各目标物的分子离子,并对分子离子进行裂解,得到二级碎片信息,优化毛细管电压、锥孔电压和碰撞能量等参数,使各化合物的响应最大化。优化后的质谱监测参数见表1。在选定的色谱和质谱条件下,混合标准溶液中各化合物的定量离子色谱图见图1。



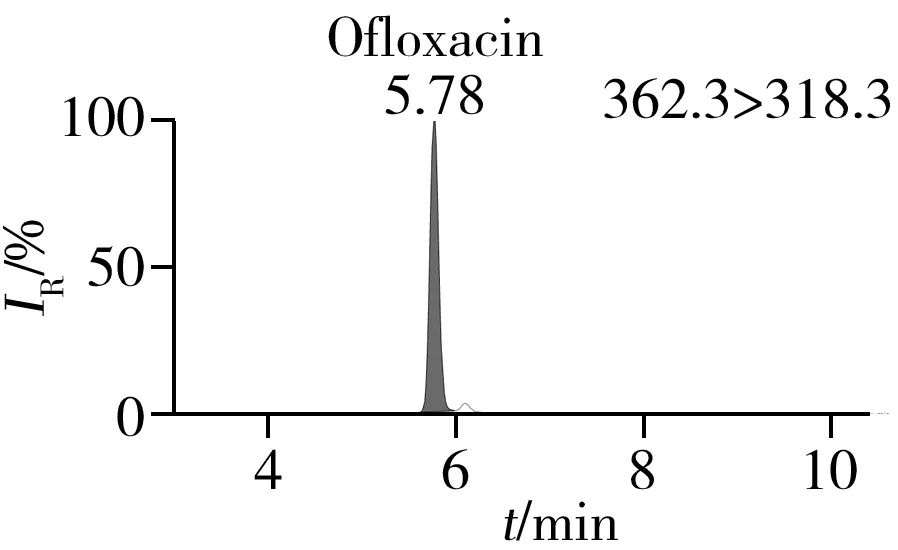



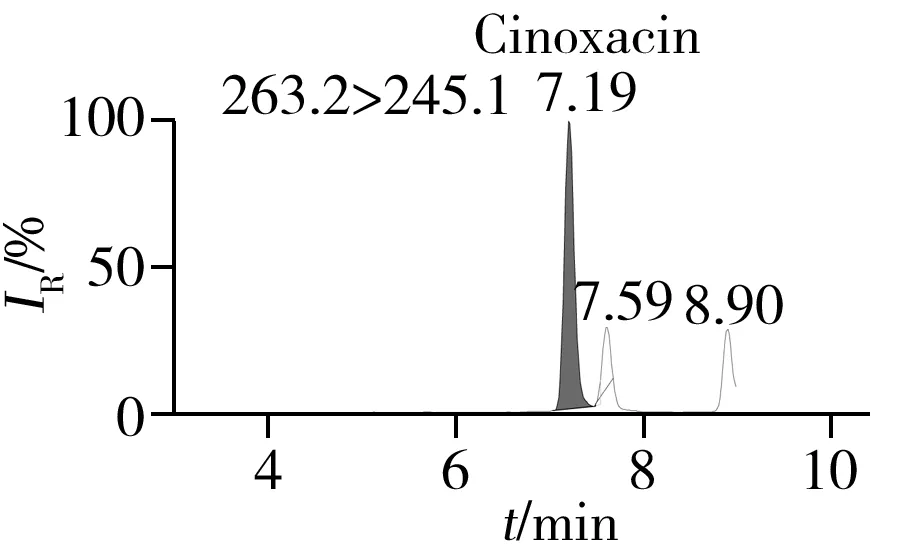





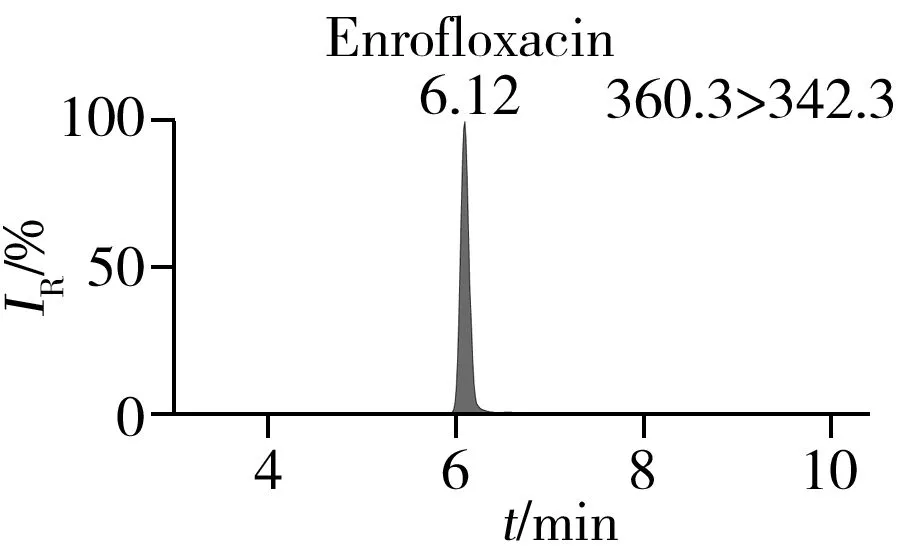




图1 混合标准样品的定量离子色谱图Fig.1 Quantitative ion chromatograms of mixed standards
2.2 提取溶剂的优化
常用的抗生素提取溶剂有乙腈、乙腈-水、甲醇和甲醇-水等,水产品中含有较多的蛋白质和脂类等大分子物质,采用甲醇或甲醇-水溶液提取,提取液较混浊,样品净化困难,且回收率偏低。乙腈具有沉淀蛋白质的作用,并且其带出的弱极性成分少,质谱响应高、基质干扰小,因而选用乙腈作为主要提取溶剂。本实验首先考察了60%乙腈水、70%乙腈水、80%乙腈水和90%乙腈水4种不同配比提取剂的提取效果,结果表明,用80%乙腈水作为提取剂时各化合物的回收率较高。分析原因,经60%乙腈水和70%乙腈水提取后的提取液较混浊,基质效应大,经80%乙腈水和90%乙腈水提取后的提取液较澄清,蛋白基本沉淀,但90%乙腈水中乙腈比例过高,使蛋白质快速凝结成团,而凝结的蛋白质阻碍了提取液进入内部发生作用,添加一定比例的水可起到分散作用。
由于喹诺酮类化合物的分子结构中含有羧基和叔胺基,具有酸碱两性,易溶于酸性或碱性溶液,在有机溶剂中添加一定比例的酸或碱提取可显著提高回收率[16-17]。考察了80%乙腈水和80%乙腈水(含0.1%甲酸)2种提取剂对回收率的影响,结果显示酸性提取剂可获得更优的回收率。因此,最终确定提取溶剂为80%乙腈水(含0.1%甲酸)。
2.3 离心条件的优化
样品经提取剂提取后,需进行离心分离得到澄清的上清液,以便进行后续的净化处理。本实验对离心条件进行了考察。结果表明,经普通离心机(<5 000 r/min)作用后,提取液仍浑浊,不利于净化,随着离心力的加大,提取液逐渐澄清,经15 000~20 000 r/min离心分离后,上清液基本澄清,过PRiME HLB时较通畅。水产品中含有较多的蛋白质和脂肪,低速离心只能分离普通的固体悬浮颗粒,而高速离心能够加快液体中颗粒的沉降速度,高速分离悬浮介质中较小的颗粒物质,从而达到理想的分离效果。本研究确定的离心速率为20 000 r/min,离心时间为5 min。
2.4 净化条件的优化
本实验采用Oasis PRiME HLB固相萃取柱对样品进行净化,考察了Oasis PRiME HLB(200 mg,6 mL)的柱容量,比较了不同上样体积的样品净化程度。结果表明,上样液中样品量≤0.5 g时,基质效应小,净化效果明显,而样品量为0.5~1.0 g时,基质效应逐渐增加,说明样品量为0.5 g时该柱对杂质的吸附能力接近饱和,过多的上样体积会造成部分杂质无法吸附,随目标化合物共流出,导致净化效果不理想。因此,本实验确定上样体积为2 mL(含0.4 g样品量)。
2.5 线性关系、检出限、定量下限与加标回收率

对阴性鱼肉样品和虾肉样品分别进行加标实验,加标量为5、10、20 μg/kg,每个浓度设6个平行,分别计算回收率和相对标准偏差(RSD),并根据信噪比,以及质谱本身的稳定性、不同仪器间的差异和方法的适用性,确定本方法的检出限(LOD)与定量下限(LOQ),结果见表2~3。结果表明,鱼肉中各目标化合物的回收率为83.4%~119.9%,RSD为 2.6%~14.9%,检出限为0.5 μg/kg;虾肉中各目标化合物的回收率为72.1%~118.3%,RSD为2.4%~15.6%,检出限为0.5 μg/kg。该方法具有良好的准确度和精密度,可以满足水产品中喹诺酮类药物的检测要求。
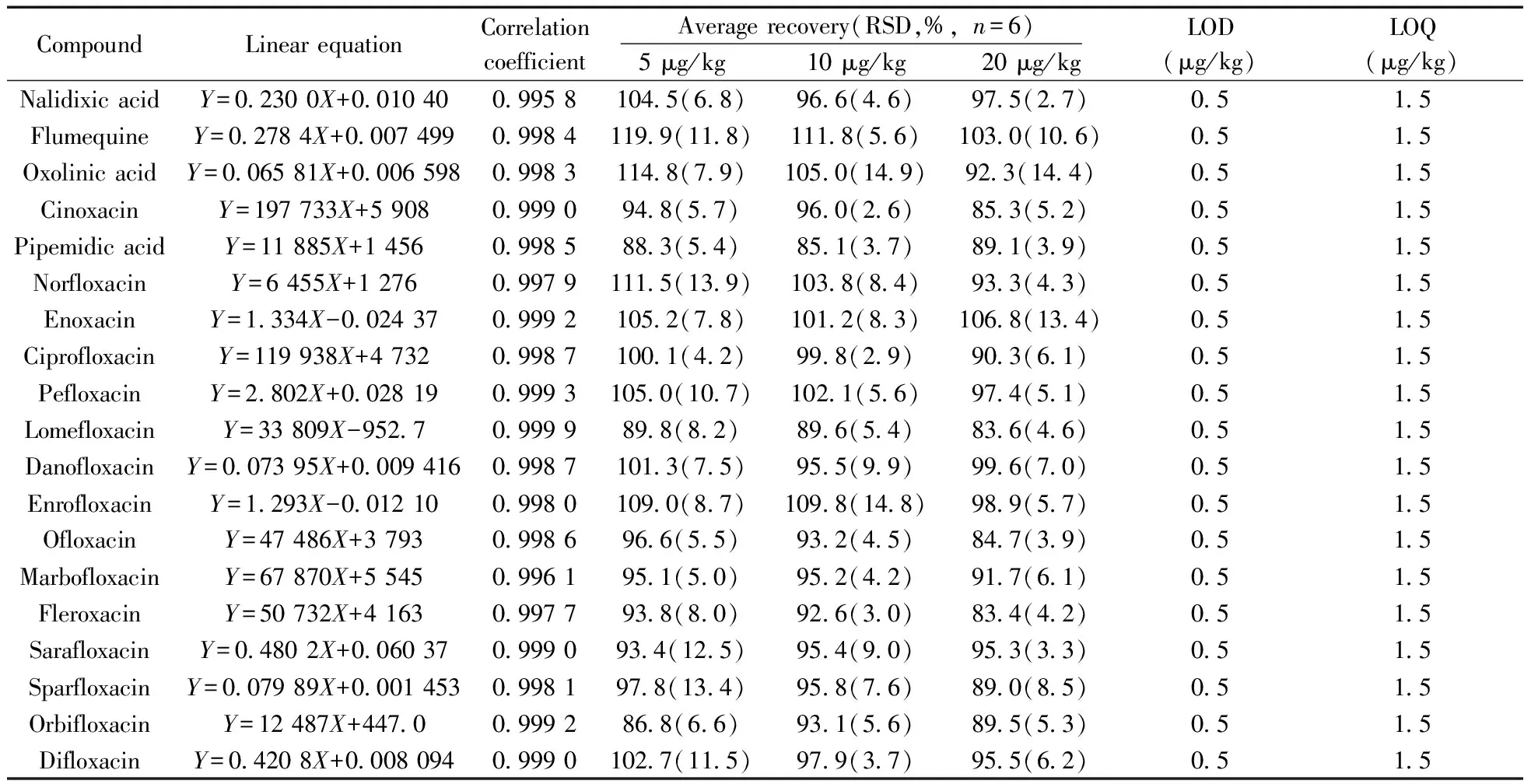
表2 鱼肉中喹诺酮类药物的线性关系、检出限、定量下限、回收率和精密度Table 2 Linear equations,correlation coefficient(r),limit of detection,limit of quantitation,recovery and precision of quinolone antibacterials in fish

表3 虾肉中喹诺酮类药物的线性关系、检出限、定量下限、回收率和精密度Table 3 Linear equations,correlation coefficient(r) ,limit of detection,limit of quantitation,recovery and precision of quinolone antibacterials in shrimp meat
2.6 实际样品的分析
采用本方法对30份市售水产品进行测定,结果在1批黑鱼中检出氧氟沙星(含量93.8 μg/kg),1批黄鳝中检出诺氟沙星(含量260 μg/kg),阳性样品的TIC色谱图见图2。根据农业部公告第2292号,我国将停止在食用性动物中使用氧氟沙星和诺氟沙星等兽药,本结果说明在水产养殖过程中乱用兽药现象依然严重,值得引起有关部门的重视。

3 结 论
本研究应用PRiME HLB净化技术,以超高效液相色谱-四极杆串联质谱联用方法建立了水产品中19种喹诺酮类药物残留的检测方法。通过对水产品中的鱼肉和虾肉进行方法学评价,证明各组分在1~20 μg/L范围内线性关系良好,平均回收率为72.1%~119.9%,RSD为2.4%~15.6%。本方法适用于水产品中喹诺酮类药物残留检测,具有准确、快速、简便、灵敏度高等优点。
[1] Li J S,Qiu Y M,Wang C.DrugResidueAnalysis.Shanghai:Shanghai Science and Technology Press(李俊锁,邱月明,王超.兽药残留分析.上海:上海科学技术出版社),2002:256-299.
[2] Domagala J M.J.AntimicrobialChemotherapy,1994,33(4):685-706.
[3] Liu L J,Liu H,Liang M,Ma S Y.Med.Herald(刘丽娟,刘虹,梁敏,马世尧.医药导报),2005,24(10):959-960.
[4] Maximum Residue Limit of Veterinary Drugs in Animal Food.Announcement No.235 of the Ministry of Agriculture(动物性食品中兽药最高残留限量.农业部235号公告),2002.
[5] Ev Lda S,Schapoval E E.J.Pharm.Biomed.Anal.,2002,27(1/2):91-96.
[6] Zheng J,Huang X R,Li Y P.FoodSci.(郑晶,黄晓蓉,李耀平.食品科学),2004,25(10):247-250.
[7] Huet A C,Charlier C,Tittlemier S A,Singh G,Benrejeb S,Delahaut P.J.Agric.FoodChem.,2006,54(8):2822-2827.
[8] Zhao S J,Jiang H Y,Li X L,Mi T J,Li C,Shen J Z.J.Agric.FoodChem.,2007,55(10):3829-3834.
[9] Kirbis A,Marinsek J,Flajs V C.Biomed.Chromatogr.,2005,19(4):259-265.
[10] Samanidou V,Evaggelopoulou E,Trotzmuller M,Guo X H,Lankmayr E.J.Chromatogr.A,2008,1203(2):115-123.
[11] Johnston L,Mackay L,Croft M.J.Chromatogr.A,2002,982:97-109.
[12] Ju L Y,Song X H,Gu J,Xu C G,Yang L J.J.Instrum.Anal.(鞠玲燕,宋晓华,谷婕,徐成钢,杨丽君.分析测试学报),2016,35(1):42-47.
[13] Wang J,Zhang X Y,Hao J,Xu B W,Chen W B.FineandSpecialtyChemicals(王骏,张秀妍,郝佳,许炳雯,陈文博.精细与专用化学品),2017,25(1):41-44.
[14] Guo M M,Li Z X,Wang Z,Pan M X,Wu H Y.J.Instrum.Anal.(郭萌萌,李兆新,王智,潘明轩,吴海燕.分析测试学报),2017,36(3):337-342.
[15] Wan Y W,Deng K G,Liu L,Huang H W.ChineseJournalofVeterinaryScience(万译文,邓克国,刘丽,黄华伟.中国兽医学报),2012,32(12):1886-1889.
[16] Cao J,Chen Y,Yang R Z,Huang Y F.ChineseJournalofVeterinaryDrug(曹军,陈勇,杨瑞章,黄月芳.中国兽药杂志),2011,45(7):21-25.
[17] Li H F,Yin J G,Liu Y M.FisheryQualityandStandardinChina(李慧芳,殷军港,刘永明.中国渔业质量与标准),2012,2(1):62-66.
