益肾补骨液高效液相色谱法特征图谱研究
2018-03-02王婷婷肖俊美王宝成
徐 云,王婷婷,肖俊美,王宝成 ,徐 建*
(1.吉林修正药业新药开发有限公司 吉林省中药标准化关键工程技术重点实验室,长春 130103;2. 通药制药集团股份有限公司,吉林 通化 134000)
益肾补骨液由骨碎补、何首乌、茯苓、续断、白芍、当归等12味中药制成。具有滋补肝肾,强壮筋骨功效。用于肝肾不足,劳伤腰痛,筋骨损伤。现行标准收载于部颁标准中药成方制剂第10册,仅有何首乌理化鉴别,何首乌、白芍薄层鉴别,无法全面表征该中药制剂的物质基础和化学成分群的整体性和复杂性,难以全面的控制益肾补骨液的内在质量。本实验研究建立了益肾补骨液HPLC特征图谱,对10批益肾补骨液进行了指纹图谱分析。益肾补骨液与组方药味间有较好的相关性,采用该法可帮助确定色谱峰的归属,进一步分析益肾补骨液的化学组成及来源,为益肾补骨液的质量控制提供理论依据[1-5]。
1 仪器与试药
1.1 仪器 安捷伦1200高效液相色谱仪;戴安(双三元)高效液相色谱仪;AB204-E型分析天平。1.2 试药 甲醇、乙腈为色谱纯,水为超纯水,磷酸等其他试剂均为分析纯;益肾补骨液(批号:160901、160902、160903、160904、160905、160906、160907、160908、160909、160910)均由通药制药股份有限公司提供;没食子酸(批号:110831-200302 )、 2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷(批号:110844-201512 )、芍药苷对照品(批号110736-201438)、柚皮苷对照品(批号:110722-201111 )、绿原酸 (批号:110753-201314 )均由中国食品药品检定研究院提供。
2 方法与结果
2.1 色谱条件[6-9]色谱柱:ACE C18色谱柱,(250 mm×4.6 mm,5 μm);流动相:以甲醇为流动相A,以0.02%磷酸溶液为流动相B,梯度洗脱(0~ 40 min,5%A → 40%A,40~ 50 min,40%A → 40%A,50~ 51 min,40%A → 5%A,51~60 min,5%A→5%A;)流速为1 mL/min;检测波长为230 nm;柱温为35 ℃,进样量:5 µL。
2.2 溶液制备
2.2.1 混合对照品溶液制备 取没食子酸、芍药苷、绿原酸、2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷、柚皮苷适量,精密称定,分别加甲醇制成每1 mL分别含0.1 mg的混合溶液,即得。
2.2.2 供试品溶液制备 取本品,精密量取15 mL,加水至30 mL。用乙醚振摇提取2次,30 mL/次,弃去乙醚液,水液用水饱和正丁醇振摇提取3次,30 mL/次,合并正丁醇液,再用正丁醇饱和水洗2次,20 mL/次,合并正丁醇液,蒸干,残渣加甲醇溶解定容至10 mL量瓶中,摇匀,滤过,取续滤液作为供试品溶液。
2.3 方法学考察
2.3.1 精密度试验 取同一份供试品溶液连续进样6次,以芍药苷为参照峰,计算各特征峰的相对保留时间,计算结果RSD值均小于2.0%。结果表明,该仪器测试精度良好。
2.3.2 重复性试验 取同一批(批号:160901)益肾补骨液6份,分别按供试品溶液制备方法制备供试品溶液,记录色谱图,以芍药苷为参照峰,计算各特征峰的相对保留时间,计算结果RSD值均小于2.0%。结果表明方法的重复性良好。
2.3.3 稳定性试验 取同一份益肾补骨液的供试品溶液,分别在0、4、8、12、16、20 h进样,记录色谱图,以芍药苷为参照峰,计算各特征峰的相对保留时间,计算结果RSD值均小于2.0%。结果表明供试品溶液在20 h内稳定。
2.4 特征图谱建立及相关性分析 取10批益肾补骨液成品,按上述选定的供试品溶液制备方法和检测方法进行检测。将10批益肾补骨液成品的测试数据导入国家药典委员会“中药指纹图谱相似度评价系统(2004A)”软件,采用中位数法,多点校正,自动匹配,共标定13个共有峰,并鉴定归属了5个特征峰;对13个特征峰进行归属:1号(没食子酸)、6号、8号(芍药苷)特征峰归属为白芍药材;2号、3号、4号(绿原酸)、5号、7号、10号特征峰归属为续断药材;9号(二苯乙烯苷)特征峰归属为何首乌药材;11号和13号(柚皮苷)特征峰归属为骨碎补药材。10批成品特征图谱叠加图、生成的对照图谱分别见图1~图2。
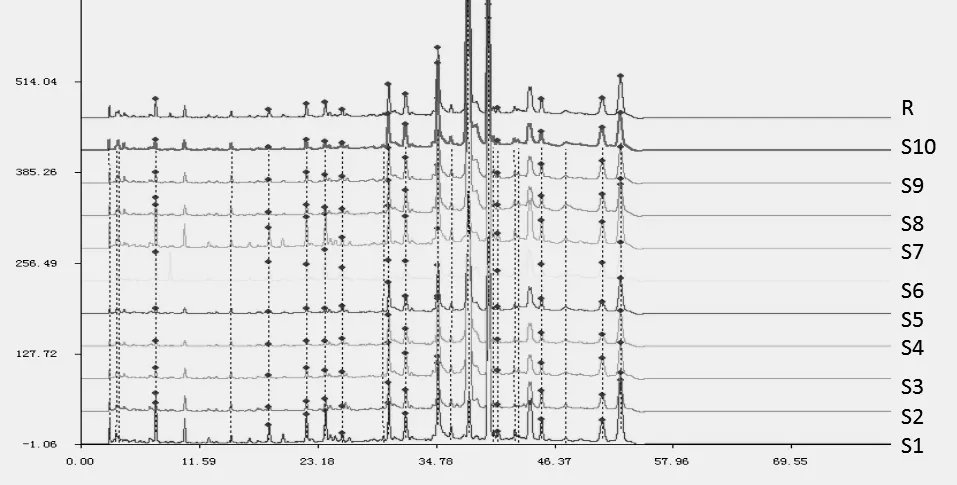
图1 10批成品特征图谱叠加图

图2 成品对照特征图谱
3 讨论
采用二级阵列检测器,对供试品检测波长进行了考察,考察200~400 nm全波长扫描,并重点考察在230、283、320 nm波长处的色谱图,结合DAD三维图及基线的波动程度、色谱峰的数目、信号响应强弱,确定230 nm 为最佳波长。考察了流动相:1)甲醇-水系统,线性洗脱。2)乙腈-水系统,线性洗脱。3)甲醇-磷酸水系统,非线性洗脱。流动相3)系统较好,并在此系统下进行了进一步的优化[10-17]。益肾补骨液主要色谱峰与其组方药味间有较好的相关性,采用该法可帮助确定药效部色谱峰的归属,进一步分析益肾补骨液化学组成及来源,可为益肾补骨液的质量控制提供理论保证。
[1]杨献玲,徐多多,高阳,等.高效液相色谱法建立黄芪多糖特征图谱研究[J].长春中医药大学学报, 2016, 32(4):701-703.
[2]徐建,徐云,王鑫,等.骨骼风痛片中鸡血藤高效液相特征图谱研究[J]. 长春中医药大学学报, 2013, 29(5):928-930.
[3]李红梅,佟志军,孙丽,等.满山红中黄酮类成分液相色谱指纹图谱研究[J].长春中医药大学学报, 2016, 32(4):708-710.
[4]魏颖,王东超,高佳琪,等.HPLC-DAD 法同时测定糖痹康中6 种主要活性成分[J].长春中医药大学学报, 2016,32(6):1135-1138.
[5]江洁怡,董玉娟,李养学,等.桂枝配方颗粒的HPLC 指纹图谱研究[J].长春中医药大学学报, 2014, 30(6):1015-1018.
[6]国家药典委员会.中华人民共和国药典[M].北京:中国医药科技出版社, 2015:105.
[7]冯冰,陈婷. HPLC测定复方昆丹胶囊中芍药内酯苷,芍药苷,二苯乙烯苷,五没食子酰葡萄糖[J].中国实验方剂学杂质, 2012, 18(7):112-116.
[8]刘岩. HPLC法测定参苏片中柚皮苷的含量[J].长春中医药大学学报, 2014, 30(3):417-427.
[9]付玉梅,张俐伟,伍振峰,等.陈皮药材水提物的HPLC特征指纹图谱研究[J].中成药, 2006, 28(1):8-10.
[10]张琦,王镇中,萧伟,等.白芍药材UPLC特征指纹图谱研究[J].中国中药杂志, 2011, 36(6):712-714.
[11]张志海,王彩云,杨天鸣,等.陈皮的化学成分及药理作用研究进展[J].西北药学杂志, 2005, 20(1):47-48.
[12]彭双,韩立峰,王涛,等.骨碎补中的化学成分及药理作用研究进展[J].天津中医药大学学报, 2012, 31(2):122-125.
[13]孙桂波,纪凤兰.何首乌的化学成分及药理作用研究进展[J].长春中医药大学学报, 2007, 23(4):105-106.
[14]彭锐,马鹏,李龙云,等.不同产地川党参药材的HPLC指纹图谱[J].中国中药杂志, 2010, 35(2):183-186.
[15]杨来英,崔芳,吴国泰,等.当归药材不同提取部分的指纹图谱分析[J].中国实验方剂学杂志, 2014, 20(13):55-60.
[16]鄢海燕,邹纯才.瓜蒌薤白颗粒的高效液相色谱特征图谱及与组方药味的相关性[J].中国医院药学杂志, 2013,33(4):276-280.
[17]王海丽,潘薇,张国跃,等.儿童回春颗粒HPLC特征图谱研究[J].药物分析杂志, 2016, 36(9):1689-1695.
