基于两种一测多评法对尾叶香茶菜中四种二萜类成分的质量控制研究
2018-02-22杨丽霞陈勇强
杨丽霞,陈勇强,孙 舰
(1.山西中医药大学,山西晋中030619; 2.晋中学院化学化工学院,山西晋中030619;3.广西中医药大学赛思斯医药学院,广西南宁530222)
尾叶香茶菜 (Rabdosia excisa(Maxim.)Hara)又名龟叶草,是唇形科香茶菜属(Rabdosia)植物,在我国的东北、日本、朝鲜以及俄罗斯远东地区都有分布[1],全草入药,具有清热解毒、健胃、活血的功效。用于治疗胃炎、膀胱胀痛、感冒发热、乳腺癌、关节痛、蛇虫咬伤等[2]。药理研究表明,尾叶香茶菜中的二萜类化合物Kamebakaurin、excisanin A、rabdokunmin C和Kamebanin对P388有明显的抑制作用[3-4]。目前,用于尾叶香茶菜药材质量控制与评价的方法主要是对尾叶香茶菜活性成分含量的精确测定[5-7],不能全面科学地控制与评价尾叶香茶菜药材质量;同时,多个活性成分的含量测定需要耗费多种对照品,而一些对照品又存在难以制备、不稳定、难以获得或价格昂贵等问题。一测多评法的应用可以解决对照品缺乏的难题。本实验采用HPLC法同时测定了尾叶香茶菜中Kamebakaurin、excisanin A、rabdokunmin C和Kamebanin的含量,并利用以上4种二萜类物质内在的函数和比例关系,采用一测多评法[8-9],以 Kamebakaurin 为内参物,计算得 excisanin A、rabdokunmin C、Kamebanin与Kamebakaurin之间的相对校正因子(fk/s),从而得出4种二萜类物质的含量,为更确切评价尾叶香茶菜药材的质量提供了新的分析模式。
1 材 料
1.1 仪器
LC-20A高效液相色谱仪,Diamonsil C18色谱柱(250 mm×4.6 mm,5 μm),Shim-pack VP-ODS C18色谱柱(250 mm×4.6 mm,5 μm),InertSustain C18色谱柱(250 mm×4.6 mm,5 μm)均为日本岛津公司;DV215CD分析天平(十万分之一,美国OHAUS公司);DV114C分析天平(万分之一,美国OHAUS公司);KQ5200V超声清洗仪(江苏省昆山市淀山湖镇)。
1.2 试 药
8批尾叶香茶菜药用植物采集于辽宁本溪、抚顺、丹东、鞍山,并按照《中国药典》2015版(一部)规定进行干燥,粉碎,过40目筛等预处理[10];对照品 Kamebakaurin、excisanin A、rabdokunmin C和Kamebanin由本实验室制备,纯度均在98%以上。乙腈(色谱纯,天津四有生物医学科技有限公司);甲醇(分析纯,天津科密欧化学试剂有限公司);纯净水(杭州娃哈哈集团有限公司)。
2 方法与结果
2.1 HPLC定量方法
2.1.1 色谱条件 色谱柱:Diamonsil C18柱(250 mm×4.6 mm,5 μm);流动相为乙腈-水(体积比23∶77);流速 1.0 mL/min;运行时间 65 min;柱温25℃;检测波长230 nm;进样量20 μL;两次进样之间用流动相平衡15 min。色谱图见图1。
2.1.2 混合对照品溶液的制备 精密称取对照品Kamebakaurin 16.82 mg、excisaninA 5.85 mg、rabdokunmin C 4.82 mg和Kamebanin 4.38 mg分别置于25 mL容量瓶中,加甲醇溶解并稀释至刻度。分别精密吸取上述溶液各1.0 mL置于同一10 mL容量瓶中,用甲醇定容,摇匀即得混合对照品溶液。
2.1.3 供试品溶液的制备 取干燥尾叶香茶菜药材均匀粉末,精密称取1.0 g,用甲醇提取3次(2 h、1.5 h、1 h),每次加甲醇 15 mL,水浴加热回流温度72℃,合并提取液,减压过滤得浸膏,加甲醇溶解浸膏并定容至100 mL,摇匀,用0.45 μm微孔滤膜过滤,取其滤液,即得。

图1 混合对照品(A)和供试品(B)HPLC图
2.1.4 线性关系考察 精密称量化合物Kamebakaurin、excisaninA、rabdokunmin C 和 Kamebanin的对照品,分别配制成浓度为0.005 mg/mL,0.015 mg/mL,0.03 mg/mL,0.06 mg/mL,0.12 mg/mL,0.24 mg/mL的单一对照品溶液,分别取上述对照品溶液20 μL 进样分析,以进样量(X,μg)对峰面积积分值(Y)进行回归,得 Kamebakaurin、excisanin A、rabdokunmin C、Kamebanin 的回归方程,见表 1,各标准曲线在线性范围(0.1~4.8 μg)内线性良好。

表1 4种成分的线性关系考察结果
2.1.5 精密度试验 精密吸取同一混合对照品溶液20 μL,连续进样6次,记录各色谱峰峰面积,计 算 得 Kamebakaurin、excisanin A、rabdokunmin C、Kamebanin峰面积的 RSD(n=6)分别为 0.73%、0.52%、0.44%、0.34%,表明仪器精密度良好。
2.1.6 稳定性试验 取干燥尾叶香茶菜药材均匀粉末(新宾8月)1.0 g,按“2.1.3”项制得供试品溶液,在0~12 h内每隔2 h进样1次,每次进样20 μL,测定峰面积。利用峰面积平均值考察样品在甲醇溶液中的稳定性,结果显示,化合物Kamebakaurin、excisanin A、rabdokunmin C、Kamebanin 在各时间点峰面积的RSD(n=6)分别为0.94%、1.12%、1.18%、1.09%,说明样品在甲醇中12 h内稳定性良好。
2.1.7 重复性试验 取干燥尾叶香茶菜药材均匀粉末(新宾8月)6份,每份 1.0 g,按“2.1.3”项制得6份供试品溶液,用0.45 μm微孔滤膜过滤,取其滤液20 μL进样分析。测定结果显示,样品中化合物 Kamebakaurin、excisanin A、rabdokunmin C、Kamebanin含量的RSD(n=6)分别为0.92%、1.17%、1.25%、1.62%,说明重复性良好。
2.1.8 加样回收率试验 取尾叶香茶菜药材均匀粉末6份,每份1.0 g,精密称定,分别向其中加入对照品 Kamebakaurin 5.40 mg、excisanin A 2.48 mg、rabdokunmin C 1.18 mg、Kamebanin 1.18 mg,按“2.1.3”项制得6份加标提取液,各取25.0 mL稀释至50 mL,得6份加标溶液。另取尾叶香茶菜药材均匀粉末6份,每份1.0 g,同“2.1.3”项制得6份供试品溶液。取供试品溶液和加标溶液分别进样分析,测定含量,计算回收率。结果化合物Kamebakaurin、excisanin A、rabdokunmin C、Kamebanin的平均回收率分别为99.66%(RSD为0.89%)、99.75%(RSD为1.97%)、99.50%(RSD 为 2.27%)、99.86%(RSD为2.01%),表明方法的准确度符合要求(n=6)。
2.1.9 耐用性试验 参照文献[11]方法,分别考察流动相配比变化±2%、柱温变化±2℃、检测波长变化±2 nm、体积流量变化±10%,以及采用3根不同色谱柱(Diamonsil C18色谱柱,Shim-pack VP-ODS C18色谱柱,InertSustain C18色谱柱)进行测定时,仪器色谱行为的变化。结果显示,不同条件下所测得四种二萜类成分含量的RSD均在2.3%以内,分离效果良好。因此,尾叶香茶菜中的四种二萜类成分测定条件较宽,具有较好的耐用性[12]。
2.2 相对校正因子(fk/s)的计算
2.2.1 多点校正法 参照文献[11-13]方法,按照公式fk/s=(Cs×Ak)/(Ck×As)计算,得到其他成分相对于Kamebakaurin的fk/s。结果见表2。同时可根据公式Ck′=(Cs×Ak′)/(fk/s×As)计算其他三种待测成分的质量浓度。
2.2.2 斜率校正法 参照文献方法[11],按照斜率校正法公式fk/s=ak/as计算(其中as为“2.1.4”项下所得标准曲线内参物斜率,ak为“2.1.4”项下所得标准曲线其他对照组分斜率),得出excisanin A、rabdokunmin C、Kamebanin相对于Kamebakaurin的 fk/s分别为0.952、0.933、1.042。同时可根据公式Ck′=Ak′/(as×fk/s)计算其他三种待测成分的质量浓度。
2.3 fk/s重现性考察
实验考察了两种仪器下3根色谱柱(Diamonsil C18色谱柱,Shim-pack VP-ODS C18色谱柱,InertSustain C18色谱柱)对fk/s的影响。结果见表3。

表2 以Kamebakaurin为参照的fk/s(多点校正法)

表3 不同仪器和色谱柱下的fk/s
2.4 待测组分的色谱峰定位
利用相对保留时间进行定位,即以Kamebakaurin为基准峰,计算不同仪器和色谱柱下excisanin A、rabdokunmin C、Kamebanin峰相对保留时间。结果见表4。
2.5 一测多评法与外标法测定结果的比较
分别精密吸取8批尾叶香茶菜供试品溶液各20 μL,进样分析,记录 Kamebakaurin、excisanin A、rabdokunmin C及Kamebanin的峰面积,用外标法和两种一测多评法分别计算四种组分的含量,结果见表5。可得,尾叶香茶菜中的4种二萜类成分在不同采收期、不同采收地含量差异较大。采用t检验法,对多点校正法与外标法得到的尾叶香茶菜药材中 excisanin A、rabdokunmin C、Kamebanin的含量进行比较,P>0.05,表明两种方法测得的含量差异无统计学意义;多点校正法与外标法所得含量间的相对标准偏差均小于5%,说明多点校正法应用于尾叶香茶菜药材的多指标成分质量评价中是可行的。采用t检验法,对斜率校正法与外标法得到的尾叶香茶菜药材中以上3种二萜类成分的含量进行比较差异有统计学意义(P<0.05),表明两种方法测得的含量差异有统计学意义;多点校正法与外标法所得含量间的相对标准偏差部分大于5%,说明斜率校正法应用于尾叶香茶菜药材的质量评价有欠妥当。
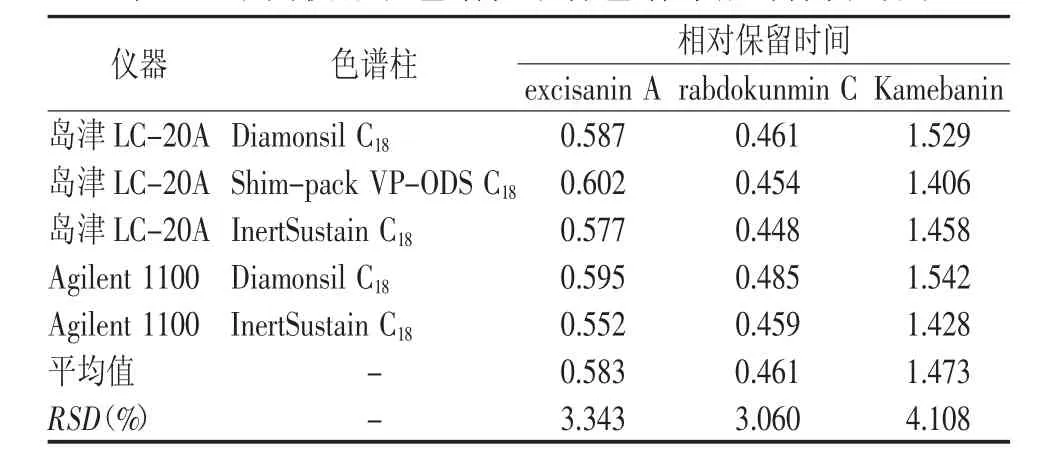
表4 不同仪器和色谱柱下各色谱峰相对保留时间
3 讨 论
考察了水、乙醇、甲醇3种提取溶剂,室温冷浸、超声提取、回流提取、索氏提取、微波提取5种提取方法,结果以甲醇加热回流提取的效率最佳,样品色谱图谱特征峰最为明显,因此本实验选用甲醇加热回流提取。
Kamebakaurin、excisanin A、rabdokunmin C 及Kamebanin是尾叶香茶菜的主要特征性成分,在药材中含量较高,药理活性显著,是评价尾叶香茶菜药材质量的适宜指标。因此实验中选取这4种化合物作为质控指标,并建立它们之间的相对校正因子fk/s。鉴于化合物Kamebakaurin在尾叶香茶菜中含量最高、易于制取、药效显著,实验中选用Kamebakaurin为内参物,建立该成分与其他3种活性成分间的相对校正因子fk/s。
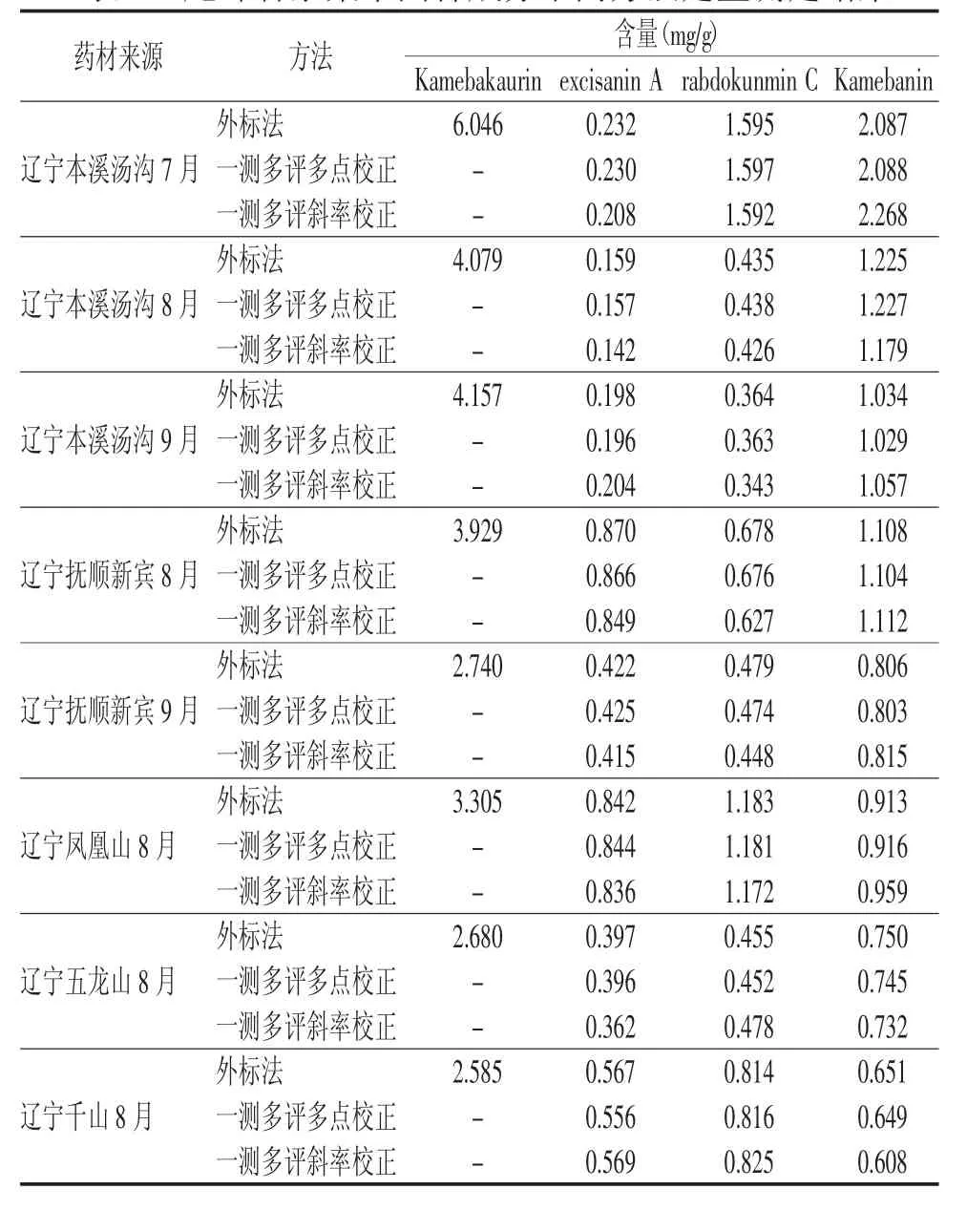
表5 尾叶香茶菜中四种成分不同方法定量测定结果
本实验首次采用两种一测多评法多点校正法和斜率校正法对尾叶香茶菜药材的4种二萜类成分同时进行含量测定。斜率校正法作为其中一种一测多评法进行待测组分含量计算时,由于不同仪器峰面积计算方式不一,使得定量因子fk/s出现一定差异,所以每台仪器的fk/s需单独建立,在评价中存在一定的局限性。从表5可得,用斜率校正法所计算的结果与外标法所得结果差异有统计学意义(P<0.05)。而多点校正法所计算的结果与外标法所得结果差异无统计学意义(P>0.05),且不同实验条件下各成分间的fk/s重现性良好(RSD<5%),说明多点校正法在对照品缺乏的前提下,可以作为一种方便、快捷、准确的方法应用于尾叶香茶菜药材多成分的含量测定和药材质量的初步评价。
实验中利用相对保留时间进行待测组分色谱峰定位(RSD<5%),但鉴于尾叶香茶菜药材色谱峰较复杂,为了保证QAMS色谱峰定位的准确性,应当结合光谱法或质谱法进行定位。
