NKCC1和KCC2 在致痫性局灶性脑皮质发育不良中表达差异性研究
2018-02-06米吉提沙克徐波涛阿吉木库尔班库尔班江努尔沙力江艾尔肯
米吉提·沙克, 徐波涛, 吕 超, 阿吉木·库尔班, 库尔班江·努尔, 沙力江·艾尔肯
(1新疆喀什地区第一人民医院神经外一科, 新疆 喀什 844000; 2南方医科大学南方医院神经外科, 广州 510515; 3第四军医大学西京医院神经外科, 西安 710043)
局灶性脑皮质发育不良(focal cortical dysplasia,FCD)是一种以局部脑皮质结构紊乱和神经元形态、功能异常为主要特征的疾病,其发生与皮层神经元发育、分化、成熟和迁移障碍相关。作为神经外科的一种非常重要的疾病,FCD的发生机制和致痫性机制一直是神经科学工作者研究的一个重要课题。研究表明,除了中枢神经递质及其受体系统功能异常外,中枢神经元氯离子通道表达异常也与FCD相关性癫痫的发生密切相关[1-2]。氯离子通道蛋白还通过对神经元内氯离子浓度的调节影响其形态塑造和功能发育[3-6]。可以说,氯离子通道表达异常是FCD发生及其致痫性机制的共同原因。近年来针对钠-钾-氯离子共同转运体1(NKCC1)和钾-氯离子共同转运体2(KCC2)在FCD致痫灶中的表达变化研究已经初步开展,但不同研究的结论并不完全一致。以往的研究大多对FCDII型病例进行探讨,而关于FCDI型病例中氯离子通道的表达变化鲜有研究,完整报道FCD不同亚型中氯离子通道表达变化的研究也未见公开报道。本研究通过Western Blot(WB)、免疫荧光和RT-QPCR方法在蛋白和mRNA 2个层面对FCD不同亚型间NKCC1和KCC2的表达差异进行探讨,并初步探讨表达差异发生的关键调控点,希望从分子层面证实证实FCD Palmini分型的意义,为进一步阐明FCD不同亚型间的临床和手术预后差异提供理论依据,并为FCD相关性癫痫的个体化治疗和分子靶向药物治疗提供新思路。
1 资料与方法
1.1一般资料收集2013年3月-2015年7月,新疆喀什地区第一人民医院和南方医科大学南方医院神经外科接受手术治并经术后病理证实为FCD的49例癫痫患者致痫灶标本进行研究,其中男性29例,女性20例,年龄27~69岁,平均年龄(40.1±1.2)岁。
1.2方法实验标本取材于新鲜的手术切除标本。依据我院及南方医科大学南方医院病理科的术后病理诊断,参考FCD Palmini分型标准,将采集实验标本分为FCD Ⅰa、Ⅰb、Ⅱa和Ⅱb 4组。设实验组和自身对照组,实验组标本取材于手术中皮层脑电监测提示的癫痫责任灶皮质,自身对照组标本取材于皮层脑电监测提示的非癫痫责任病灶皮质。实验标本大小约1 cm×1 cm,需要整块取材并取全皮质全层,用10%中性福尔马林溶液固定,包埋、切片、荧光染色。免疫荧光染色在定性层面探索NKCC1和KCC2在FCD不同亚型间的表达及其差异性。用于WB的实验标本在手术切除标本上进行3个不同区域的重复取材,需要取全皮质全层但不包含脑白质,取材标本剪碎混匀后立即于-196℃液氮中冻存,在-80℃冰箱中长期保存,一抗浓度1∶500。以灰度值检测NKCC1和KCC2的蛋白的相对表达水平。RT-QPCR的实验标本取材和保存方法与WB的标本相似,但需要去RNA酶的冻存管分装标本;经GeneBank(NCBI)查出NKCC1和KCC2蛋白的mRNA序列,内参选取ACTB;引物由Primer 5.0自动分析和设计(表1)。通过熔解曲线图形状和Tm值判断扩增特异性;扩增效率判定根据扩增曲线计算内参基因ACTB的CT值,内参基因CT值在17~30之间可以接受。
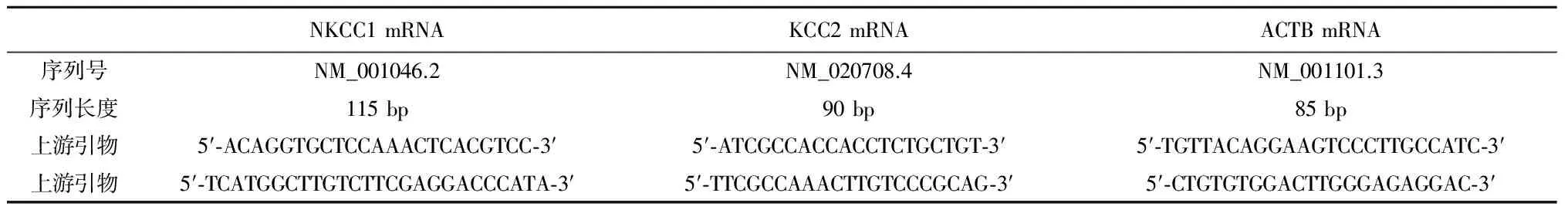
表1 RT-QPCR引物设计和合成
2 结果
2.1一般情况病例分组和纳入情况:49例病例共分为4组,包括FCD Ⅰa(10例),FCD Ⅰb(20例),FCD Ⅱa(9例),FCD Ⅱb(10例)4组。WB实验研究中共纳入47例,其中2例FCD Ⅰb亚型病例由于内参灰度值差异过大未被纳入。RT-QPCR实验研究中49例病例全部纳入。
2.2NKCC1蛋白和KCC2蛋白表达情况在FCD Ⅰa和Ⅰb亚型中,NKCC1在实验组中的相对表达量与自身对照组比较差异无统计学意义(P>0.05);在FCD Ⅱa和Ⅱb亚型中,NKCC1在实验组中的相对表达量与自身对照组比较出现上调,差异有统计学意义(P<0.05)(图1~2、表2)。进一步应用两独立样本T-Test比较,NKCC1在2组中的上调强度差异有统计学意义(P<0.05)。
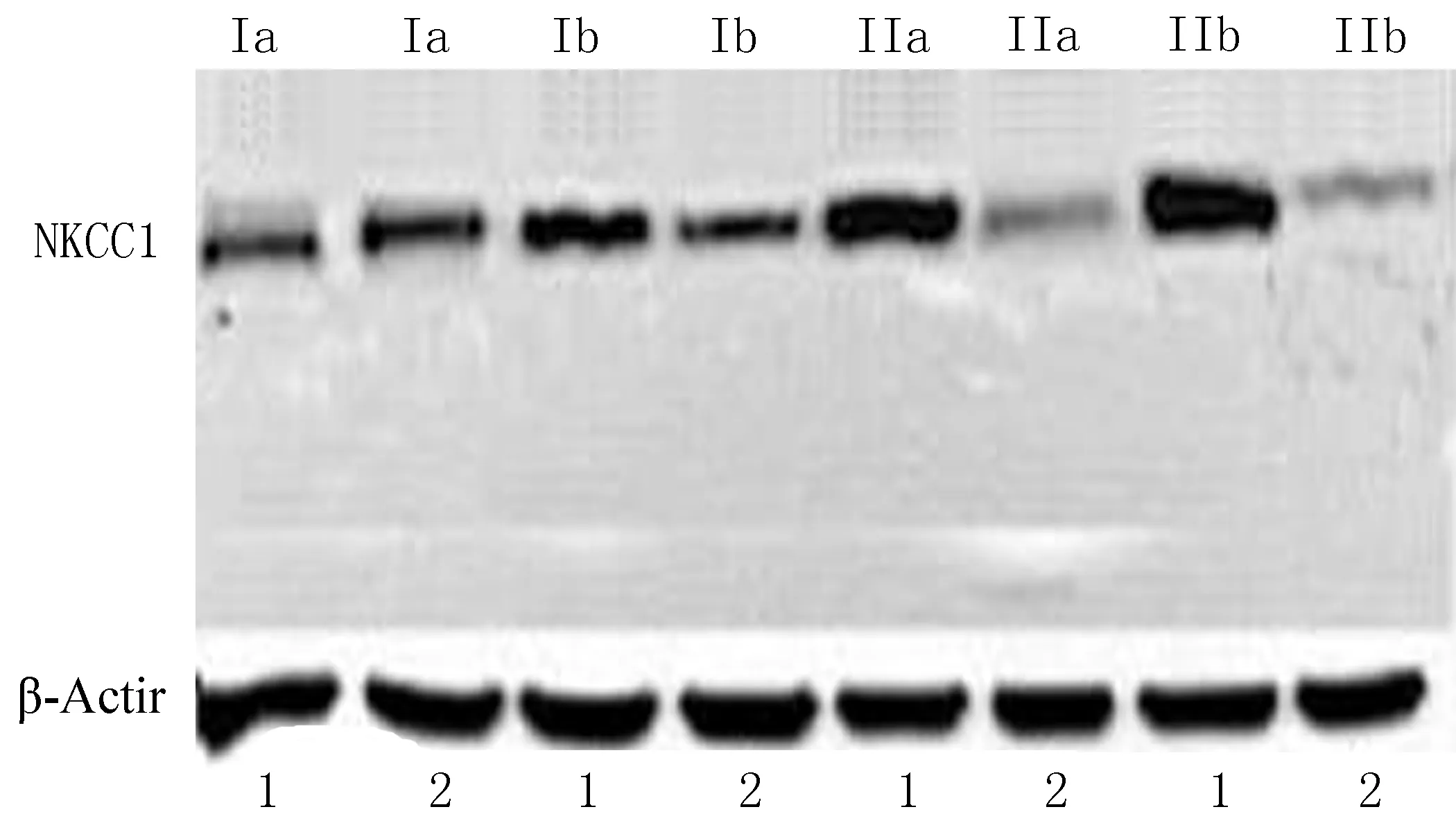
(1: 实验组; 2: 自身对照组)
在FCD Ⅰa、Ⅰb和Ⅱb亚型中,KCC2 在实验组中的相对表达量低于自身对照组,出现表达下调,差异有统计学意义(P<0.05);FCD Ⅱa 亚型中,KCC2在实验组和自身对照组中的相对表达量无统计学差异(P>0.05)。进一步应用One-way ANOVA 比较了KCC2在FCD Ⅰa、Ⅰb 和Ⅱb 中的表达下调程度差异。KCC2在FCD Ⅰa、Ⅰb 和Ⅱb 中相对表达量倍比均值分别为0.529、0.889、0.588,表达下调程度差异有统计学意义(P=0.000)。基于LSD 方法进行多重比较,FCD Ⅰa和FCD Ⅰb亚型中,KCC2下调程度差异有统计学差异(P=0.000);FCD Ⅰa和FCD Ⅱb亚型中,KCC2下调程度无统计学意义(t=28.997,P>0.05);FCD Ⅰb 和FCD Ⅱb两组中KCC2下调程度差异统计学意义(P<0.05)。
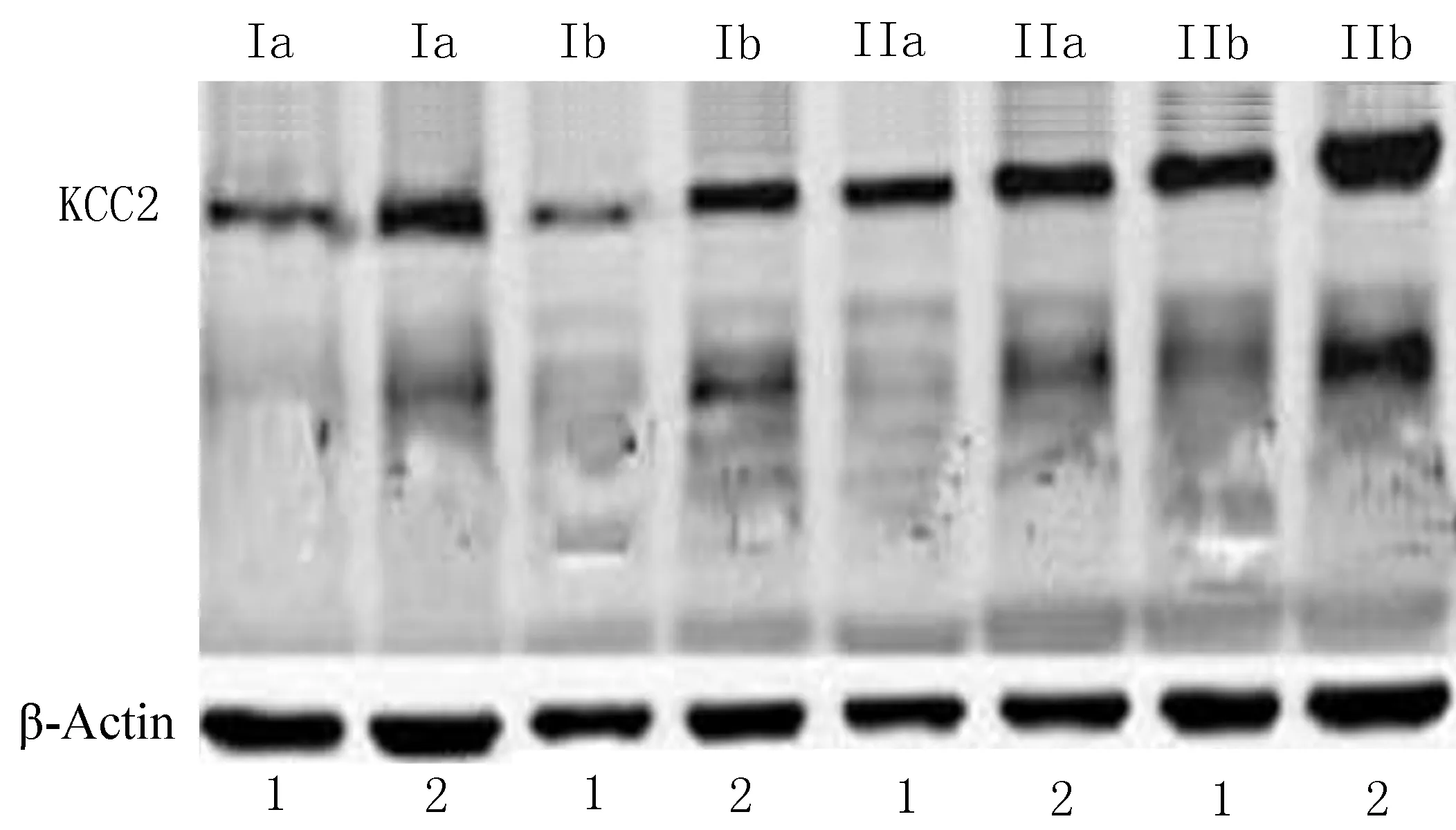
(1: 实验组; 2: 自身对照组)
2.3NKCC1mRNA和KCC2mRNA表达量在FCD Ⅰa和Ⅰb亚型中,NKCC1 mRNA在实验组中的相对表达量与自身对照组比较差异无统计学意义(P>0.05);在FCD Ⅰa亚型中,KCC2 mRNA 在实验组中的相对表达量与自身对照组相比出现上调,差异有统计学意义(P<0.05);在FCD Ⅰb亚型中,KCC2 mRNA 在实验组中的相对表达量与自身对照组比较出现下调,差异有统计学意义(P<0.05)。在FCD Ⅱa和Ⅱb 两组中,NKCC1 mRNA 在实验组中的相对表达量与自身对照组比较出现上调,差异有统计学意义(P<0.05);在 FCD Ⅱa 组中,KCC2 mRNA 在实验组中的相对表达量与自身对照组比较差异无统计学意义(P>0.05);在 FCD Ⅱb 组中,KCC2 mRNA 在实验组中的相对表达量与自身对照组相比出现上调,差异有统计学意义(P<0.05)(表3)。
3 讨论
近年来,CCC家族的表达异常与皮质发育不良性疾病(包括结节性硬化、神经节细胞瘤、FCD等)的关系越来越受到人们的重视。药物难治性癫痫是包括FCD在内的一大类皮质发育不良性疾病最常见的临床表现,其分子基础是皮质神经元分化、发育、成熟和迁移障碍。隶属于CCC家族的NKCC1和KCC2在皮质神经元中的发育性表达变化,对于神经元内氯离子浓度调节有着极为重要的作用。而氯离子浓度的发育性变化与皮质神经元发育、成熟、形态塑造、神经突触联系建立和神经元迁移等方面关系密切[3-6],因此氯离子通道的表达异常在这类疾病发生中所起的作用是目前该领域的研究热点,以氯离子通道和其调节通路为靶点的新型抗癫痫药物开发也有很大的应用前景。
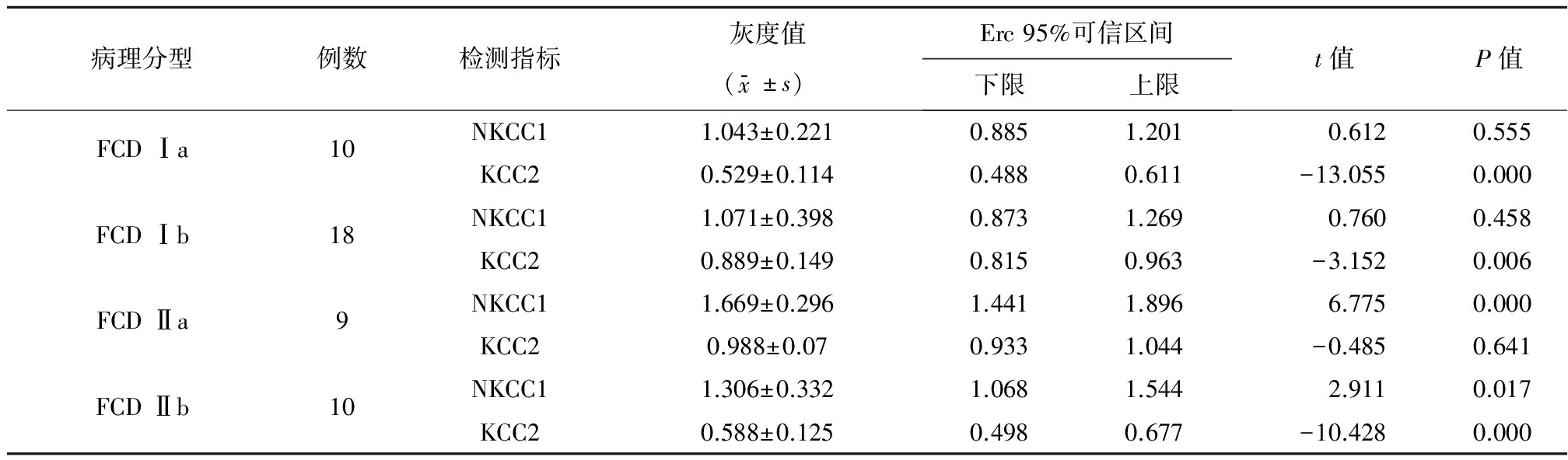
表2 NKCC1和KCC2相对表达量灰度值分析结果

表3 NKCC1 mRNA和KCC2 mRNA 相对表达量CT值分析结果
NKCC1和KCC2功能的发挥受多层次、多因素的综合和调控。总体而言,CCC功能调节包括快速调节和慢速调节过程。快速调节过程多发生在正常的生理状态下和应激病理状态下,主要通过细胞激酶介导的通道蛋白氨基末端的苏氨酸磷酸化和去磷酸化过程进行[7-9];慢速调节多涉及基因转录和转录后层面的调节,这种机制的异常与皮质发育不良性疾病、神经痛、神经退行性变等疾病密切相关[10]。
本研究基于FCD Palmini分型,在蛋白和mRNA层面对NKCC1和KCC2在FCD不同亚型之间的表达差异进行研究。研究发现,只有FCDⅡb亚型同时涉及了NKCC1和KCC2 2种蛋白的表达异常,而其他3种亚型中仅单一涉及NKCC1或KCC2的表达异常;在蛋白表达调节层面,转录层面的调节在NKCC1的表达异常中较为重要,而转录层面和转录后层面的调控在KCC2的表达异常中同时发挥了重要作用。
在NKCC1的表达变化研究中,NKCC1仅在FCDⅡ型(包括FCDⅡa和Ⅱb)中出现表达上调,表达上调出现在包括“气球样”神经元(FCDⅡb亚型)在内的所有皮质神经细胞,而FCDⅠ型中NKCC1 的表达基本正常。此前关于FCD中NKCC1 表达变化的研究全部是关于FCDⅡ型的[1,11-12],结果与本研究基本一致,而针对FCDⅠ型中NKCC1的表达变化尚无研究报道。Sen等[1]研究指出“气球样”神经元中NKCC1表达缺如,而Aronica等[12]的研究结果则提示畸形神经元和“气球样”神经元中均存在NKCC1的表达上调。本研究结论与Aronica等[12]一致,在“气球样”神经元中也存在NKCC1表达上调,程度与畸形神经元类似。在KCC2的表达变化研究中,除FCD Ⅱa亚型中表达基本正常,其他3种亚型实验组中KCC2均出现了不同程度的表达下调。此前关于FCD中KCC2表达变化的研究基本是关于FCDⅡ型的[11-15],其中KCC2在FCD Ⅱb亚型中表达下调的研究结论较一致,本研究也取得了类似的结论,但FCDⅡa亚型中KCC2表达变化的研究结论并不一致,部分研究提示该亚型中KCC2表达亦下调[13-15],而最近Talos等[11]得出了FCDⅡa中KCC2表达上调的结论,并认为其表达上调可能是防止癫痫进展的一种代偿性结果。但关于为什么其代偿仅出现在FCDⅡa亚型中并未给出合理的解释。本研究结果提示KCC2在FCD Ⅱa亚型中的表达未出现明显下降,但也未观察到其代偿性的表达上调。
在NKCC1 mRNA的表达变化研究中,NKCC1 mRNA仅在FCD Ⅱ型(包括FCDⅡa和Ⅱb)中出现表达上调,而FCDI型中NKCC1 mRNA的表达基本正常。此前关于FCD中NKCC1 mRNA表达变化的研究极少,并且未见关于人类FCD致痫中表达变化的报道,Shimizu-Okabe等[20]对皮质发育不良模型小鼠进行研究,发现病灶处NKCC1 mRNA表达出现上调。本研究结果中NKCC1 mRNA的表达变化与蛋白的表达变化一致,这说明NKCC1的表达的调控主要在转录水平发挥作用,转录后水平的调控作用相对比较弱。
在KCC2 mRNA的表达变化研究中,KCC2 mRNA在FCDⅠa和Ⅱb亚型中表达上调,在FCDⅠb亚型中表达下调,而在FCDⅡa中,表达未见明显变化。Shimizu-Okabe等[20]曾发现皮质发育不良模型小鼠病灶处KCC2 mRNA表达下调,而在其另一篇关于人FCD致痫灶的研究中,Shimizu-Okabe报道KCC2 mRNA在FCD中整体出现表达下调,而存在畸形神经元的亚型中下调程度更大。与此前的报道不同,本研究发现KCC2 mRNA在FCDⅠa和Ⅱb亚型中表达上调,与蛋白的表达变化并不一致。KCC2 mRNA出现表达上调说明KCC2的表达受到了表达后多层次的反馈和调控,慢性调控机制导致代偿性的蛋白mRNA表达量增加,以弥补蛋白表达的不足,同时可能还伴有KCC2蛋白功能和活性的上调。至于KCC2 mRNA为何只在上述两个亚型中出现上调,这可能与KCC2蛋白表达在这2个亚型中表达下调程度有关,WB结果提示KCC2在FCDⅠa和Ⅱb亚型中表达下调程度明显大于FCDⅠb亚型,而蛋白表达不足的代偿和调节机制启动可能与蛋白表达下调的强度有关。总之,本研究结果提示KCC2 mRNA的表达变化与蛋白的表达变化并不完全一致,这说明KCC2的表达的调控不仅在转录水平发挥作用,同时也在转录后水平发挥作用。
[1] 吴政. 氯离子共转运体KCC2在癫痫发病机制中的作用及其作为抗癫痫靶点的研究[D]. 复旦大学, 2011.
[3] 张慈柳, 彭镜, 何芳,等. CIC-3氯离子通道蛋白在内侧颞叶癫痫中的表达变化[C]// 中华医学会第十七次全国儿科学术大会论文汇编(下册). 2012.
[3] GE S, GOH E L, SAILOR K A, et al. GABA regulates synaptic integration of newlygenerated neurons in the adult brain[J]. Nature, 2006, 439(7076): 589-593.
[4] CANCEDDA L, FIUMELLI H, CHEN K,et al.Excitatory GABA action is essential formorphological maturation of cortical neurons in vivo[J]. J Neurosci, 2007,27(19): 5224-5235.
[5] REYNOLDS A, BRUSTEIN E, LIAO M,et al. Neurogenic role of the depolarizingchloride gradient revealed by global overexpression of KCC2 from the onset ofdevelopment[J]. J Neurosci, 2008, 28(7): 1588-1597.
[6] LI H, KHIRUG S, CAI C,et al.KCC2 interacts with the dendritic cytoskeleton topromote spine development[J]. Neuron, 2007, 56(6): 1019-1033.
[7] PALMINI A, NAJM I, AVANZINI G,et al. Terminology and classification of thecortical dysplasias[J]. Neurology, 2004, 62(6 Suppl 3):S2-S8.
[8] ADRGNA N C, DI FULVIO M, LAUF P K. Regulation of K-Cl cotransport: from function to genes[J]. J Membr Biol, 2004, 201(3): 109-137.
[9] FLATMAN P W. Regulation of Na-K-2Cl cotransport by phosphorylation andprotein-protein interactions[J]. Biochim Biophys Acta, 2002, 1566(1-2):140-151.
[10] KAHLE K T, STALEY K J, NAHED B V, et al. Roles of the cation-chloridecotransporters in neurological disease[J]. Nat Clin Pract Neurol, 2008,4(9):490-503.
[11] TALOS D M, SUN H, KOSARAS B, et al. Altered inhibition in tuberous sclerosis andtype IIb cortical dysplasia[J]. Ann Neurol, 2012, 71(4):539-551.
[12] ARONICA E, BOER K, REDEKER S,et al.Differential expression patterns of chloridetransporters, Na+-K+-2Cl-cotransporter and K+-Cl-cotransporter, in epilepsy-associated malformations of cortical development[J]. Neuroscience, 2007,145(1): 185-196.
[13] SHIMIZU-OKABE C, TANAKA M, MATSUDA K, et al. KCC2 was downregulated insmall neurons localized in epileptogenic human focal cortical dysplasia[J].Epilepsy Res, 2011, 93(2/3): 177-184.
[14] ROBINSON S, MIKOLAENKO I, THOMPSON I, et al. Loss of cation-chloridecotransporter expression in preterm infants with white matter lesions:implications for the pathogenesis of epilepsy[J]. J Neuropathol Exp Neurol,2010, 69(6):565-572.
[15] JANSEN L A, PEUGH L D, RODEN W H, et al. Impaired maturation of cortical GABA(A) receptor expression in pediatric epilepsy[J]. Epilepsia, 2010,51(8):1456-1467.
[16] WANG C, SHIMIZU-OKABE C, WATANABE K,et al.Developmental changes in KCC1,KCC2, and NKCC1 mRNA expressions in the rat brain[J]. Brain Res Dev Brain Res, 2002, 139(1):59-66.
[17] BEN-ARI Y, KHALILOY I, KAHLE KT,et al.The GABA excitatory/inhibitory shift inbrain maturation and neurological disorders[J]. Neuroscientist, 2012,18(5):467-486.
[18] CONTI L, PALMA E, ROSETI C,et al.Anomalous levels of Cl-transporters cause adecrease of GABAergic inhibition in human peritumoral epileptic cortex[J].Epilepsia,2011, 2(9):1635-1644.
[19] TAYLOR D C, FALCONER M A, BRUTON C J, et al. Focal dysplasia of the cerebral cortexin epilepsy[J]. J Neurol Neurosurg Psychiatry, 1971, 34(4):369-387.
[20] SHIMIZU-OKABE C, OKABE A, KILB W, et al. Changes in the expression ofcation-Cl-cotransporters, NKCC1 and KCC2, during cortical malformationinduced by neonatal freeze-lesion[J]. Neurosci Res, 2007, 59(3):288-295.
