单个等离子体纳米颗粒在生化分析和生物成像中的应用
2018-01-29雷刚何彦
雷刚,何彦
1 引言
众所周知,等离子体纳米颗粒(PNPs)因其独特的局域表面等离子共振(LSPR)吸收和散射而展现出优良的光学性质1。近年来,PNPs的光散射性质得到了越来越多的关注,并逐渐被应用于分析化学领域2。在光散射分析化学的初期,研究者们主要利用溶液中大量纳米颗粒的平均散射信号来进行定量分析3,4,尽管这种分析方法快速、简便且检测范围广泛,但其在灵敏度和准确度方面必然存在一定的局限性。暗场显微镜(DFM)的出现促进了单个 PNPs的 LSPR光散射性质方面的研究,也为实现单纳米颗粒水平上的分析提供了可能5-7。PNPs的LSPR散射光对周围介质环境非常敏感,有机小分子和生物大分子的吸附以及化学反应都能够引起PNPs表面介质环境的变化,从而使其 LSPR散射光谱发生相应的移动,因此,单个 PNPs可以作为彼此独立的光散射探针用于分子检测以及其它相互作用的分析。
作为一种具有超高灵敏度的检测技术,单分子光谱(SMS)技术自被建立以来就受到大量关注,并于近十年间得到迅猛的发展8,为分析化学开启了一扇全新的大门。SMS技术是考察复杂细胞系统内动力学变化及物质相互作用的强有力方法。不同于以大量分子长时间内的平均行为为信号来源的传统分析方法,SMS技术可以实现对溶液中单个分子的检测与实时成像,从而排除宏观测量对系统复杂性造成的简化,发现隐藏在整体平均中的个体差异,捕捉淹没于宏观测量的极少数的中间体或过渡状态,追踪被掩盖在平衡状态之下随时间和空间变化着的微观动力学过程。目前,相对成熟的SMS技术大都是以荧光分子或者半导体量子点为探针的荧光成像技术9,如激光共聚焦、全内反射荧光等等。但是,传统的荧光小分子和蛋白光稳定性差,且光强度较弱,而半导体量子点又存在有细胞毒性较大且难以进行特定修饰等缺点10。这些问题一方面推动着开发新型荧光探针的研究,另一方面促进着非荧光标记单分子成像技术的建立。与荧光探针相比,PNPs易于修饰、生物兼容性好,且其 LSPR散射光具有强度高、稳定性好及持续性长等优点。因此,发展以PNPs为探针的单分子成像技术对化学传感、生物成像与生物纳米技术的重要性不言而喻。
单个PNPs的成像主要基于其LSPR吸收和散射特性,如微分干涉相差(DIC)成像和暗场成像。由于DIC图像对比度不高,从中难以区分探针和各种亚细胞结构,所以大多数关于成像单个PNPs的报道都是以暗场成像为手段的。暗场成像技术是一种以所测样品的散射光为信号在较低背景下进行成像的技术,通过与光谱仪联用,该技术还可用于获取单个颗粒的LSPR光谱11。目前,常见的DFM主要利用高数值孔径聚光镜产生斜照明,在空间上实现对入射光与所测样品散射光的信号分离,从而产生极高的对比度。用暗场成像技术拍摄的细胞图像背景暗样品亮,其对比度甚至可以与荧光图像媲美。但是,传统的DFM也存在分辨率不高、光学切割能力较弱和背景干扰大等缺点。通过对光源、检测器及其它光学元件的择优组装和调试,我们开发出了一系列具有高灵敏度、高时空分辨率和高通量的暗场成像技术,并采用经可控合成筛选出的单分散单个PNP探针,将这些技术巧妙地应用于单分子检测、多颗粒传感、生物过程示踪以及单细胞成像等领域。除此之外,我们还以具有光学各向异性的PNPs为探针,研制出精确的活细胞三维扫描成像系统和激光暗场高速毛细管电泳联用系统,为高效率的仪器偶联提供参照。这些技术有望将单分子光谱技术的发展推向一个全新的高度。

2 PNPs的合成
PNPs的LSPR性质与其自身的组成成分、大小和形状等有着密切的关系,为了筛选出合适的PNP探针,已有许多研究深入探讨了各种 PNPs的可控合成方法,如利用DNA介导及电置换反应等12-14。由于银和金纳米颗粒(AgNPs和 AuNPs)的吸收和散射信号主要集中在近紫外-可见-近红外光区,且强度较高,所以其已成为最常用的PNP探针15,16。相对于 AgNPs,AuNPs因为具有更稳定的光学性质和更低的细胞毒性而在生物成像中得到广泛的应用。金纳米棒(AuNRs)是最为典型的一个例子,其不仅能够产生强而稳定的散射信号,而且具有起源于自身各向异性的光学偏振特性,因此,AuNRs作为一种非常理想的光学成像探针近年来得到研究者们的广泛关注。AuNRs最早的制备是在含有十六烷基三甲基溴化铵(CTAB)的金离子溶液中通过硅或铝的微孔中经电化学还原所实现的17,此方法得到的AuNRs虽有着一些特殊的光学特性,但是没有明显的 LSPR性质。2001年,Murphy课题组最先提出晶种生长法的概念18,他们将小 AuNPs作为晶种加入到含有氯金酸和CTAB的混合溶液中,通过多步生长法得到了长径比可控的AuNRs19。然而,该方法的产率较低,而且样品需要分离,较为繁琐。在2003-2004年,很多研究小组研发了一种高产率的小长径比AuNPs的合成方法,即使用外部有1.5 nm的CTAB保护的 AuNPs为晶种,并加入一定量的 AgNO3作为辅助20-22。该方法可以通过控制硝酸银的量来调控AuNRs的长径比,所制备的AuNRs的产率在 95%以上。由于操作简单、重复性高且易于调控,这种银离子辅助的晶种生长法是目前使用最多的AuNRs制备方法。鉴于要经常使用单分散且均一的AuNRs作为生物成像和系统性能测试的探针,我们基于晶种生长法系统地研究了AuNRs的合成,发现pH值不仅能改变还原剂抗坏血酸的还原能力,还能改变CTAB分子在金表面的吸附能力以及CTAB双分子层胶束的形状和大小,并最终改变合成产物的形状23。在此基础上,我们又发现除了常用的抗坏血酸,亚硝钠和氯化铁在酸性条件下都可以作为温和的还原剂对AuNRs的长径比进行精确的调控24,相较于可控合成,这种利用刻蚀反应调节AuNRs的LSPR性质以得到满足条件的探针的方法更为简便。此外,我们还发现在碱性条件下用双氧水作还原剂可以合成小尺寸的AuNRs,弥补了在碱性条件下合成AuNRs的空缺25。由于无论采用哪种方法合成AuNRs都不可避免地会得到包含有各种长径比的AuNRs与具有其它形状的AuNPs的混合物,为了获得单分子成像必需的均一的AuNRs探针,我们发展了一种简便的、基于梯度分离法分离纯化AuNRs的新方法26。
在诸多 PNPs之中,AuNPs的生物兼容性最好,但是其它金属也有着金所不具备的优异性能,比如银的氧化活性,铂的催化性能等27,28。因此,合成具有核壳结构的金与其它金属的复合纳米材料可能会降低其它金属的细胞毒性,从而使其特殊的性能可以在应用于生物体内的研究体系中得到施展,我们课题组也开展了一些这方面的工作。最近的研究表明,活性氧系列分子(ROS)的生成与累积能够引起细胞的氧化性损伤,这也是紫外线照射引起细胞损伤的主要途径之一。因此,清除细胞内的ROS以防止其累积对维护人体健康和防治疾病有着重要的意义。为了达到清除细胞内ROS的目的,我们制备了一种表面带有刺状铂纳米点的AuNRs,相较于的单纯的铂纳米颗粒,这种复合纳米材料表现出更好的生物兼容性和催化性能,实验结果证明其能有效地催化分解细胞内的ROS从而缓解紫外线辐射所造成的细胞损伤29。我们还合成了金银复合PNPs以实现细胞内的H2S传感和调节探针颜色响应区间的目的,详细的内容见下一节。
3 PNPs在生化分析中的应用
3.1 单分子检测
由于在临床诊断方面具有潜在的应用价值,基于 PNPs聚集引起光谱变化所建立的特定核酸序列的检测和定量方法已经受到了大量的关注。如Alivisatos等所展示的那样30,通过金―硫键把两段核苷酸序列分别修饰到AuNPs表面之后,将含有与这两段核苷酸序列互补的目标 DNA溶液与之混合,DNA的三明治杂交会使得AuNPs形成二聚体或者多聚体,从而允许研究者们通过AuNPs紫外吸收光谱的位移或者肉眼可分辨的颜色变化实现对DNA的检测。这种比色检测DNA的方法虽然简便,但是存在灵敏度不高、难以定量和无法同时实现多靶物检测等缺点。本课题组使用一个带有真彩 CCD的 DFM,以单个颜色编码的AuNPs为探针,发展了一种基于耦合颗粒计数的DNA检测方法,这种方法能够对多种目标分子同时进行动态检测(图1(a, b),而且灵敏度极高,检测下限达到0.02 pmol·L-1(图1(c))31。但是,由于该方法需要将PNP探针固定在玻片上来进行计数,较为耗时,因此我们在后续工作中使用一个高亮度短脉冲的频闪光源来代替显微镜标配的卤钨灯,利用改造后的 DFM (FLDM)对溶液中的AuNPs进行直接快速的计数,通过DNA杂交引起的AuNPs探针数量的减少与目标DNA浓度之间的线性关系实现了对目标DNA的检测,从而解决了上述问题32。但是,依靠探针数量减少的方法在目标分子浓度很低时会存在较大的误差。考虑到球形的PNPs是各向同性的,经线性偏振光照射之后,不能够改变线性偏振光的振动方向,因此在起偏器与检偏器垂直的情况下,检测器无法收集到球形PNPs的光学信号。当球形PNPs发生聚集之后,其聚集体很不均一,具有很强的各向异性,因此能够产生偏振散射信号。通过在之前的FLDM 上装配起偏器和检偏器,我们发展了一种基于频闪光源的正交偏振DFM方法,由于检测的是目标分子导致的可检测探针颗粒的增加,大大提高了检测的精密度33。该方法对硫化物有着0.1 nmol·L-1的检测下限,且动态范围跨越五个数量级。

图1 利用颜色编码的PNPs建立单分子检测方法31Fig.1 Single molecule detection using color coded PNPs31.
3.2 多颗粒传感
由于PNPs的LSPR散射光对自身性质及周围环境的变化都非常敏感,因此发展能够快速且高通量地获取单个纳米颗粒的 LSPR散射光谱的技术是非常必要的。传统的基于光谱仪的单颗粒光谱成像技术通量较低,只能获取狭长区域内少数颗粒的光谱,因为其必需要使用入射狭缝来排除背景光谱的干扰。在实现了高信背比单颗粒散射光成像的基础上,我们通过去除入射狭缝,在显微镜检测光路中加入透射光栅的方法,建立了一种能够同时获取多个单颗粒的散射光谱的技术。利用该技术,我们以单个AuNRs为探针实时观察了H2O2氧化金的反应,并且通过实时分析多个纳米颗粒的动态散射光谱,发现金纳米棒在反应中会以自催化的方式加快反应的速率(图2)34。
细胞内环境复杂,经常需要同时检测不同区域的单颗粒光谱来监测细胞内某些物质的区域浓度。作为一种特殊的气体信号分子,H2S在许多生物系统中具有重要的调控作用。采用这种高通量的单颗粒暗场光谱成像技术,我们以具有核壳结构的银包金纳米棒为探针,对细胞内硫化物的局部浓度进行了高灵敏地检测。我们的方法基于对因形成硫化银所导致的纳米探针 LSPR光谱红移的实时监测。特别地,我们发现单个金银复合纳米探针的光谱的变化与局部硫化物浓度以及在细胞内环境复杂,经常需要同时检测不同区域的单颗粒光谱来监测细胞内某些物质的区域浓度。作为一种特殊的气体信号分子,H2S在许多生物系统中具有重要的调控作用。采用这种高通量的单颗粒暗场光谱成像技术,我们以具有核壳结构的银包金纳米棒为探针,对细胞内硫化物的局部浓度进行了高灵敏地检测。我们的方法基于对因形成硫化银所导致的纳米探针 LSPR光谱红移的实时监测。特别地,我们发现单个金银复合纳米探针的光谱的变化与局部硫化物浓度以及在单个金纳米棒表面生成硫化银的反应速率直接相关,从而首次以 nmol·L-1灵敏度实现了针对活细胞内局域硫化物水平实时变化的高灵敏度、高选择性和大动态范围的检测(图 3(a-e)35。虽然采用透射光栅能够一次性地获得多个纳米颗粒的光谱,但是提取这些光谱却要耗费大量的时间,为此,我们建立了一种通过跟踪直观易确认的颜色变化来更为简便地获取多个颗粒的光谱变化信息的方法。由于颜色响应的敏感区间主要位于可见光黄绿光区(530-590 nm),我们通过用金银核壳球形纳米颗粒取代金银核壳棒状纳米颗粒,把纳米颗粒探针的响应区间调整至颜色检测最敏感的区间,从而能够利用探针的颜色变化同时监测大量颗粒对局域H2S的响应,实现了更为简便的高通量的多颗粒硫化物传感(图3(f-h)36。根据颜色的形成机制,颜色并非一种可以测量的客观物理量,而是一种主观感觉。尽管人们能够对事物的颜色形成共识,但同一物体采用同一检测器,其颜色表现都会因照明光源的不同而产生显著差异。通过激光照明工程技术,采用多束窄带光源的组合,我们发展了基于彩色CCD三基色分光的PNPs散射光检测技术。该技术可以把颜色分辨能力提高一个数量级,使光谱差异较小、在白光下无法区分的AuNPs呈现出明显的颜色差异,从而实现纳米金粒径以及团聚解团聚的高灵敏可视化检测37。

图2 实时监测单个AuNRs的氧化动力学过程34Fig.2 Real-time monitoring the dynamic oxidation process34.
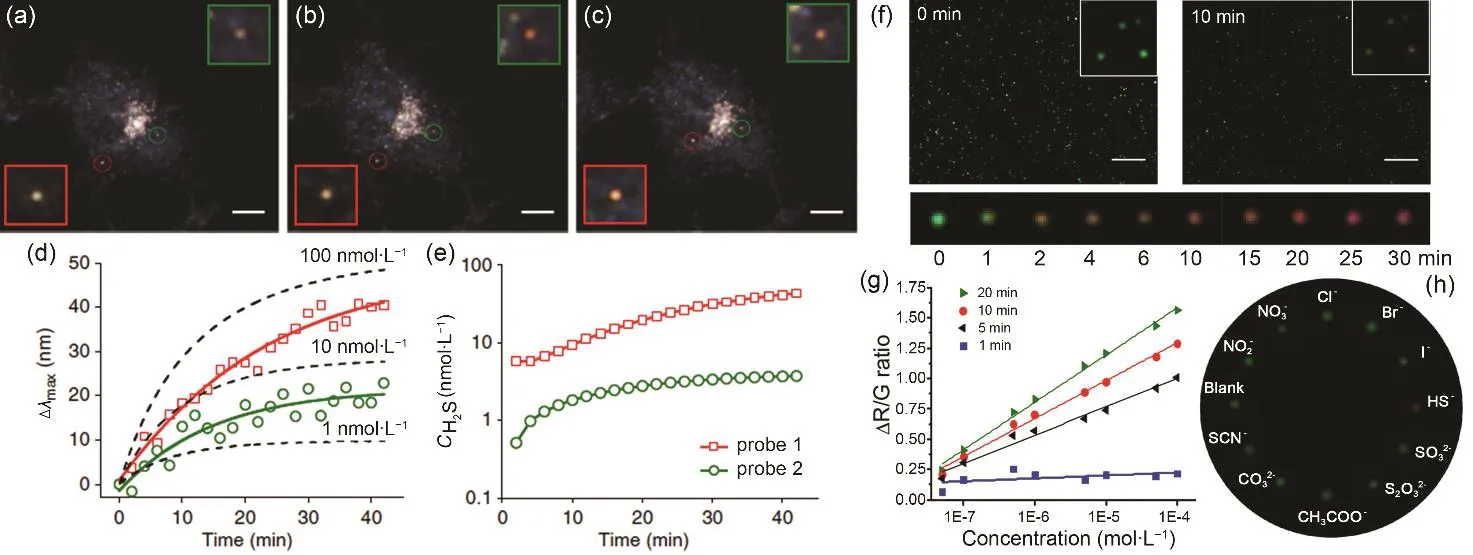
图3 体内外的高通量硫化物传感35,36Fig.3 High-throughput sulfide sensing in vitro and in vivo35,36.
4 基于PNPs的单细胞成像
当PNPs应用于单细胞成像时,由于细胞内环境复杂,存在极大的背景干扰,因此如何提高成像的信噪比以及如何区分纳米颗粒是在细胞膜上还是细胞内部一直是一个亟待解决的问题。此外,尽管 PNPs常被用作光散射探针以实时示踪细胞内的一些生命活动和代谢过程38,39,但是其进入细胞的途径及原理一直以来都未得到进一步的揭示。本课题组长期开展了有关细胞内纳米颗粒高分辨成像以及纳米颗粒与细胞相互作用方面的研究,在此过程中研发出不少能够示踪纳米颗粒在运动过程中的三维角度变化、实现突破衍射极限的超分辨成像和活细胞三维扫描成像等技术,这些先进的成像系统将为单细胞成像方面的研究提供有力的帮助。
4.1 基于双偏振成像检测单个AuNR的三维取向
要考察单个纳米颗粒在液固表面的运动行为,不仅需要实时跟踪单个纳米颗粒的平移运动,还需要准确地知晓它们的三维角度,而传统的暗场成像技术显然不能提供这些信息。为此,我们发展了基于双通道偏振成像检测单个AuNR三维空间取向的技术。该技术的原理是把具有特定传播方向的入射光照射到含有AuNRs的样品上,由于AuNRs的长轴方向与入射光之间存在着可用数学方程准确描述的平面角与极化角,其产生的散射光信号号会同时包含具体的三维角度信息。通过在检测光路中加入一块双折射晶体,就可以同时获取该AuNR在两个正交偏振方向上的散射光总强度和相对强度,从而推算出其平面角与极化角的大小。我们通过理论和实验证明平面照明模式和环形照明模式都可以获取AuNRs的三维空间取向。采用该技术,我们考察了单个AuNR跨越细胞膜进入细胞的全过程,获得了该重要生物学行为的大量细节信息(图 4)40。此外,我们还考察了牛血清蛋白修饰的单个 AuNR与 C18修饰的玻片表面在色谱流动相淋洗过程中的相互作用,发现二者之间不是简单的吸附与解吸附的关系,而是包含了许多复杂的动力学状态(图5)41。
4.2 单个细胞的高分辨成像及三维扫描成像
有多种方法可以实现三维荧光成像,但其前提都是在接近零背景的情况下对荧光探针分子进行选择性的识别42。但是,由于检测波长与照射波长相同,以AuNPs为探针的共振光散射成像不能使用波长滤光片来消除背景瑞利散射的干扰。我们开发了一种双波长差异成像方法来解决这个问题43。如图6(a, c)所示,在波长分别为473 nm和532 nm的激光照射下,纯的细胞溶解产物的双通道图像几乎没有表现出强度的变化。相比之下,纯的AuNPs的双通道图像则展现出很大的强度变化(图6(b, d)。基于此,我们可以通过双波长成像模式下强度差异的大小来有效地区分活细胞中的细胞背景与AuNP探针,实现对单个小AuNP探针的高灵敏检测(图6(e, f)。
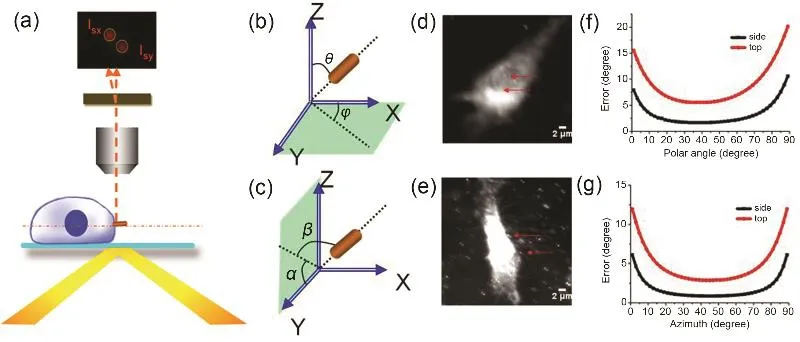
图4 直接成像单个AuNR的跨膜动力学过程40Fig.4 Direct imaging of transmembrane dynamics of single gold nanorod40.

图5 直接观察液固表面上单个蛋白质包被的纳米颗粒的方向动力学41Fig.5 Direct observation of the orientation dynamics of single protein-coated nanoparticles at liquid/solid interfaces41.

图6 活细胞中PNPs的无背景双波长差异成像43Fig.6 Noise-free dual-wavelength difference imaging of plasmonic resonant nanoparticles in living cells43.
为了实现高分辨暗场成像,我们还采用正交偏振光检测技术,基于AuNRs的LSPR散射光强度对其空间取向的敏感性建立了对各向异性AuNPs进行准确定位和超分辨成像的新方法。正交偏振成像可以有效地消除来自球形纳米颗粒、玻璃基底和细胞内部的瑞利散射光背景(图7)。我们证明该技术可实现对单个AuNRs 15 nm的定位精度和水平方向上最小80 nm左右的分辨率44。除此之外,当采用高数值孔径的物镜镜头时,通过使用一个步进旋转电机来精确地控制 Z方向上的距离,该技术还可以对单个AuNRs进行选择性地三维成像,从而实现细胞内AuNR探针三维分布的高分辨定位45。
4.3 探究单个PNPs与细胞外环境的相互作用
采用传统的单分子暗场成像技术,我们实时原位观察了50-130 nm的AuNPs与癌细胞的相互作用。在AuNPs被癌细胞内吞的过程中,它们在以扩散的方式到达细胞膜之前经历了一个明显的减速过程。通过光学切割,我们发现这层能够减慢AuNPs运动的细胞外周介质厚度大约为3-20 μm46。进一步的研究表明,这个以前被人们所忽略的、在光学显微镜下很难观察到的介质层其实是以多糖聚合物透明质酸为主要成分的细胞膜外基质(PCM),它是细胞个体的缓冲区,对细胞外纳米颗粒的捕获和内吞有重要影响。当PCM存在时,AuNPs被细胞捕获和内吞的数量较多;但当PCM被溶解掉之后,AuNPs被细胞捕获和内吞的数量就相对较少(图8)。通过对单个纳米颗粒的实时示踪,我们发现PCM的主要作用是“缓冲减速”,即把在一定尺寸范围内(50-200 nm)的AuNPs的扩
散速率,都调整到与细胞膜上蛋白受体的扩散速率相接近的水平,即0.1 μm2·s-1,从而有利于受体与AuNPs的结合,这表明PCM似乎具有某种程度的主动性。不仅如此,我们还发现能够抑制细胞内吞纳米颗粒的条件和试剂也同时会抑制PCM对纳米颗粒的捕获。所有这些结果意味着细胞内吞纳米颗粒这一重要生理功能是由以细胞膜为核心,包括细胞骨架和PCM在内的多种细胞内外结构共同完成的,是亚细胞层面上的一层精巧的自组织行为,这对于我们加深对细胞整体功能的认识具有深刻的意义47,48。
4.4 活细胞中机械力转导过程的可视化

图7 基于各向异性PNPs的亚衍射极限成像44Fig.7 Subdiffraction-limited imaging based on anisotropic PNPs44.
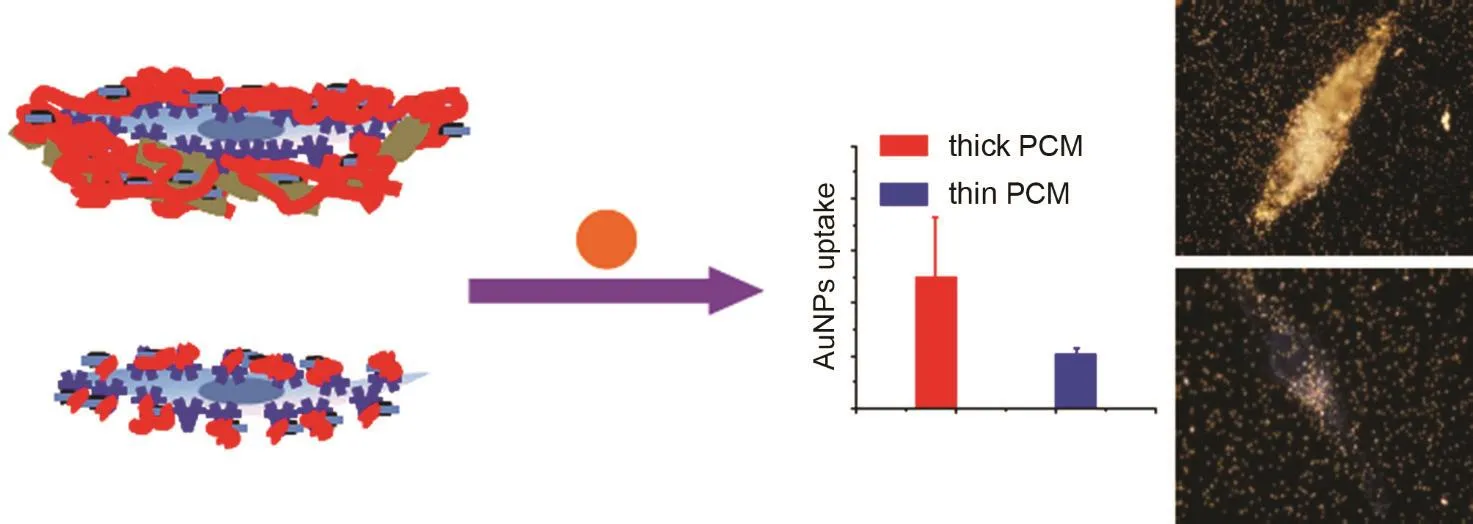
图8 细胞膜外基质对纳米颗粒滞留和摄取的增强效应47Fig.8 Pericellular matrix enhances retention and cellular uptake of nanoparticles47.
活细胞中的机械力信号被视为诸多生理功能的生物学基础,想要更好地理解这些过程的本质,就要对活细胞内的局域机械力转导的动力学过程进行成像和测定。通过合成以弹性分子PEG连接的两个金纳米颗粒构成的纳米弹簧为探针,我们利用暗场光谱成像技术对纳米弹簧因受细胞骨架的收缩或伸展的影响而产生的光谱变化进行实时监测,从而可视化了活细胞中由外加刺激信号触发的细胞膜上局域机械力的转导活动49。如图9(ac)所示,纳米弹簧的一端与玻片底部相连,另一端通过抗体与细胞膜上特定受体相连,当细胞因外部刺激而产生不均匀的收缩或伸展时,一对纳米弹簧中两个纳米颗粒之间的距离也会随之改变,从而导致两者之间的等离子耦合效应的强度变化,根据纳米弹簧LSPR散射光谱变化的大小就可以得出实时的生物机械力大小。图9(d)展示了在ROS刺激下活细胞中不同区域和不同时间段内的机械力信号变化。我们预期该策略也适用于探测活体组织中其它方式的机械力信号传导与调节。
5 单层照明暗场成像与微通道检测联用系统的构建
我们在传统的暗场及光片照明显微镜的基础上,通过对光源、检测器以及其它光学元件的选择和优化,开发出一套具有高灵敏度、高信噪比、高时空分辨率以及高各向异性分辨率的激光单层照明暗场成像体系,如图10所示。整个显微系统通过直接利用超连续白光激光光片照射样品的方式来进行暗场散射成像,由此获取纳米探针的数目、散射光颜色、光谱和强度等信息,为单个颗粒的实时动态监测提供了一种全新的研究方法。比如,利用所搭建的激光单层照明暗场成像系统,结合毛细管-缺口管阵列进样系统,我们考察了单个AuNRs在氧化过程中的动力学行为,通过分析其电迁移率、散射光颜色、光谱和强度等变化信息的变化趋势,我们发现表面带有不同修饰物的AuNRs的氧化行为之间的差异以及AuNRs在氧化过程中表面所带的电荷总量的变化,进一步地揭示了AuNRs氧化的反应机理50。

图9 活细胞中局域机械力转导的可视化49Fig.9 Visualization of localized mechanical force transduction in live cells49.
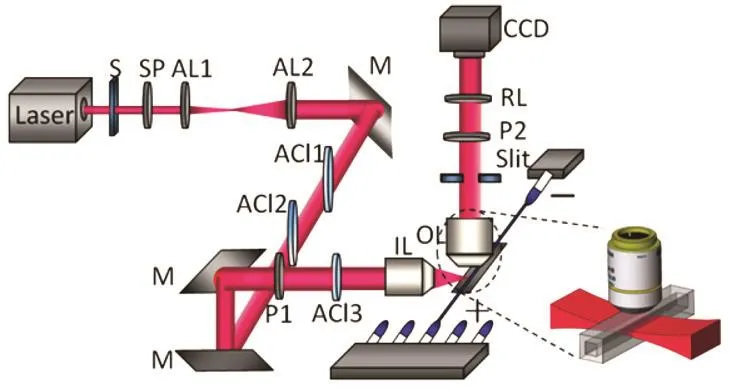
图10 用于毛细管电泳检测的超连续激光单层照明暗场成像系统的搭建原理50Fig.10 Schematic of the supercontinuum laser light-sheet plasmonic imaging system for capillary electrophoresis detection50.
6 总结与展望
PNPs因为具有优异的光学性质、催化性能和生物兼容性而逐渐从基础科学研究走向实际应用之中,其在功能材料、化学传感、生物医药和光学成像等领域已经得到了广泛的应用。PNPs的LSPR响应对周围环境极其敏感,这也使得我们能够利用其消光或散射光谱的变化对一些有害的物质进行检测。值得注意的是,暗场成像技术的应用和优化在最佳条件下可使视野中的每一个PNP都能够作为单独的传感器,这种以单个PNPs的LSPR散射光为信号的分析手段因具有常规方法难以企及的灵敏度和高通量而备受关注,具有非凡的发展空间。现有的荧光探针,包括最近兴起的碳纳米点,都存在光稳定性差或光信号较弱等缺点,相比之下,PNPs的散射光强度很高且非常稳定,更重要的是每一个PNPs都可以作为单独的探针而对样品的不同区域进行成像,使得我们能够实时原位长时间地观测同一样品中的多个区域。因此,基于PNPs的单分子光谱成像技术必将在复杂体系的成像领域中得到越来越多的重视。特别地,在近来非常活跃的活体组织和智能材料研究中,单个PNP的大小相比于材料自身的尺寸基本上可以忽略不计,从而更能够客观地反映出材料内部的结构和功能信息。此外,目前常用的单颗粒PNP探针多为金银纳米颗粒或其复合材料,很少有研究用到其它PNPs,因此该领域还有很大的发展前景,采用除金银纳米颗粒之外的单颗粒PNP探针将能进一步地拓宽单颗粒光散射成像技术的应用范围。另一方面,目前的单颗粒暗场成像技术虽能实时记录多个PNP探针的动态行为,对样品的多个区域进行同时监测,但尚不能够对每次观测获得的海量数据进行全面快速的分析,主要还是依赖研究者的个人经验选取个别有代表性的颗粒进行繁琐的手动分析,从提取的有限信息中推断出普遍性的结论。但是,随着大数据分析技术的发展和普及,我们有望利用先进的数据挖掘技术,对每一单颗粒探针的时空轨迹进行详尽的分析,并通过数据可视化技术,快速准确地从大量探针的各种时空变化过程之中提取出所需要的信息。因此,将单颗粒检测技术与大数据分析相结合,必然能够实现对复杂的生命过程更为细致的研究。总之,PNPs在生化分析和生物成像等领域仍旧有着巨大的发展潜力,而单个PNP光散射探针的应用也为单分子光谱技术注入了一股新鲜的活力。我们有理由相信,PNPs将会在未来更广泛地应用于现实生活中快速、简便的分析与成像之中。
(1) Willets, K. A.; Duyne, R. P. V. Annu. Rev. Phys. Chem. 2007, 58,267. doi: 10.1146/annurev.physchem.58.032806.104607
(2) Huang, C. Z.; Li, K. A.; Tong, S. Y. Anal. Chem. 1996, 68, 2259.doi: 10.1021/ac9511105
(3) Qi, W. J.; Wu, D.; Ling, J.; Huang, C. Z. Chem. Commun. 2010, 46,4893. doi: 10.1039/C0CC00886A
(4) de la Rica, R.; Stevens, M. M. Nat. Nanotechnol. 2012, 7, 821.doi: 10.1038/nnano.2012.186
(5) Mock, J. J.; Barbic, M.; Smith, D. R.; Schultz, D. A.; Schultz, S.J. Chem. Phys. 2002, 116, 6755. doi: 10.1063/1.1462610
(6) McFarland, A. D.; Van Duyne, R. P. Nano Lett. 2003, 3, 1057.doi: 10.1021/nl034372s
(7) Liu, G. L.; Yin, Y.; Kunchakarra, S.; Mukherjee, B.; Gerion, D.; Jett,S. D.; Bear, D. G.; Gray, J. W.; Alivisatos, A. P.; Lee, L. P.; Chen, F.F. Nat. Nanotechnol. 2006, 1, 47. doi: 10.1038/nnano.2006.51
(8) Hofkens, J.; Verheijen, W.; Shukla, R.; Dehaen, W.; De Schryver, F.C. Macromolecules 1998, 31, 4493. doi: 10.1021/ma980346i
(9) Chen, H.; Puhl, I. I. I. H. L.; Ikeda, S. R. J. Biomed. Opt. 2007, 12,054011. doi: 10.1117/1.2799171
(10) Hola, K.; Zhang, Y.; Wang, Y.; Giannelis, E. P.; Zboril, R.; Rogach,A. L. Nano Today 2014, 9, 590. doi: 10.1016/j.nantod.2014.09.004
(11) Li, Y.; Jing, C.; Zhang, L.; Long, Y. T. Chem. Soc. Rev. 2012, 41,632. doi: 10.1039/C1CS15143F
(12) Lan, X.; Chen, Z.; Liu, B. J.; Ren, B.; Henzie, J.; Wang, Q. Small 2013, 9, 2308. doi: 10.1002/smll.201202503
(13) Skrabalak, S. E.; Au, L.; Li, X.; Xia, Y. Nat. Protoc. 2007, 2, 2182.doi: 10.1038/nprot.2007.326
(14) Hu, F.; Zhang, Y.; Chen, G.; Li, C.; Wang, Q. Small 2015, 11, 985.doi: 10.1002/smll.201401360
(15) Hsu, C. W.; Zhen, B.; Qiu, W.; Shapira, O.; DeLacy, B. G.;Joannopoulos, J. D.; Soljačić, M. Nat. Commun. 2014, 5, 3152.doi: 10.1038/ncomms4152
(16) Haiss, W.; Thanh, N. T.; Aveyard, J.; Fernig, D. G. Anal. Chem.2007, 79, 4215. doi: 10.1021/ac0702084
(17) C. Hulteen, J.; Martin, C. R. J. Mater. Chem. 1997, 7, 1075.doi: 10.1039/A700027H
(18) Jana, N. R.; Gearheart, L.; Murphy, C. J. Chem. Commun. 2001, 617.doi: 10.1039/B100521I
(19) Jana, N. R.; Gearheart, L.; Murphy, C. J. J. Phys. Chem. B 2001, 105,4065. doi: 10.1021/jp0107964
(20) Eustis, S.; El-Sayed, M. A. Chem. Soc. Rev. 2006, 35, 209.doi: 10.1039/B514191E
(21) Jain, P. K.; Huang, X.; El-Sayed, I. H.; El-Sayed, M. A. Acc. Chem.Res. 2008, 41, 1578. doi: 10.1021/ar7002804
(22) Murphy, C. J.; Sau, T. K.; Gole, A. M.; Orendorff, C. J.; Gao, J.;Gou, L.; Hunyadi, S. E.; Li, T. J. Phys. Chem. B 2005, 109, 13857.doi: 10.1021/jp0516846
(23) Cheng, J.; Ge, L.; Xiong, B.; He, Y. J. Chin. Chem. Soc. 2011, 58,822. doi: 10.1002/jccs.201190128
(24) Jia, Y.; Xiong, B.; Xu, D.; Yeung, E. S.; He, Y. J. Chin. Chem. Soc.2014, 62, 141. doi: 10.1002/jccs.201400172
(25) Xu, D.; Mao, J.; He, Y.; Yeung, E. S. J. Mater. Chem. C 2014, 2,4989. doi: 10.1039/C4TC00483C
(26) Xiong, B.; Cheng, J.; Qiao, Y.; Zhou, R.; He, Y.; Yeung, E. S.J. Chromatogr. A 2011, 1218, 3823.doi: 10.1016/j.chroma.2011.04.038
(27) Lei, G.; Gao, P. F.; Yang, T.; Zhou, J.; Zhang, H. Z.; Sun, S. S.; Gao,M. X.; Huang, C. Z. ACS Nano 2017, 11, 2085.doi: 10.1021/acsnano.6b08282
(28) Fürstner, A.; Davies, P. W. Angew. Chem. Int. Ed. 2007, 46, 3410.doi: 10.1002/anie.200604335
(29) Xiong, B.; Xu, R.; Zhou, R.; He, Y.; Yeung, E. S. Talanta 2014, 120,262. doi: 10.1016/j.talanta.2013.12.020
(30) Sassolas, A.; Leca-Bouvier, B. D.; Blum, L. J. Chem. rev. 2008, 108,109. doi: 10.1021/cr0684467
(31) Xiao, L.; Wei, L.; He, Y.; Yeung, E. S. Anal. Chem. 2010, 82, 6308.doi: 10.1021/ac101018v
(32) Yuan, Z.; Cheng, J.; Cheng, X.; He, Y.; Yeung, E. S. Analyst 2012,137, 2930. doi: 10.1039/C2AN16171K
(33) Peng, L.; Cao, X.; Xiong, B.; He, Y.; Yeung, E. S. Chem. Commun.2016, 52, 7616. doi: 10.1039/C6CC02536F
(34) Cheng, J.; Liu, Y.; Cheng, X.; He, Y.; Yeung, E. S. Anal. Chem. 2010,82, 8744. doi: 10.1021/ac101933y
(35) Xiong, B.; Zhou, R.; Hao, J.; Jia, Y.; He, Y.; Yeung, E. S. Nat.Commun. 2013, 4, 1708. doi: 10.1038/ncomms2722
(36) Hao, J.; Xiong, B.; Cheng, X.; He, Y.; Yeung, E. S. Anal. Chem.2014, 86, 4663. doi: 10.1021/ac500376e
(37) Cheng, X.; Dai, D.; Yuan, Z.; Peng, L.; He, Y.; Yeung, E. S. Anal.Chem. 2014, 86, 7584. doi: 10.1021/ac501448w
(38) Chen, Z.; Li, J.; Chen, X.; Cao, J.; Zhang, J.; Min, Q.; Zhu, J. J.J. Am. Chem. Soc. 2015, 137, 1903. doi: 10.1021/ja5112628
(39) Zhang, L.; Li, Y.; Li, D. W.; Jing, C.; Chen, X.; Lv, M.; Huang, Q.;Long, Y. T.; Willner, I. Angew. Chem. Int. Ed. 2011, 50, 6789.doi: 10.1002/anie.201102151
(40) Xu, D.; He, Y.; Yeung, E. S. Anal. Chem. 2014, 86, 3397.doi: 10.1021/ac403700u
(41) Xu, D ; He,.Y ; Yeung, E. S. Angew. Chem. Int. Ed. 2014, 126, 7071.doi: 10.1002/ange.201400025
(42) Rosen, J.; Brooker, G. Nat. Photonics 2008, 2, 190.doi: 10.1038/nphoton.2007.300
(43) Xiao, L.; Wei, L.; Cheng, X.; He, Y.; Yeung, E. S. Anal. Chem. 2011,83, 7340. doi: 10.1021/ac2012366
(44) Cheng, X.; Dai, D.; Xu, D.; He, Y.; Yeung, E. S. Anal. Chem. 2014,86, 2303. doi: 10.1021/ac403512w
(45) Cheng, X.; Cao, X.; Xiong, B.; He, Y.; Yeung, E. S. Nano Res. 2017,10, 1423. doi: 10.1007/s12274-017-1524-4
(46) Zhou, R.; Xiong, B.; He, Y.; Yeung, E. S. Anal. Bioanal. Chem. 2011,399, 353. doi: 10.1007/s00216-010-4340-1
(47) Zhou, R.; Zhou, H.; Xiong, B.; He, Y.; Yeung, E. S. J. Am. Chem.Soc. 2012, 134, 13404. doi: 10.1021/ja304119w
(48) Xu, R.; Xiong, B.; Zhou, R.; Shen, H.; Yeung, E. S.; He, Y. Anal.Bioanal. Chem. 2014, 406, 5031. doi: 10.1007/s00216-014-7877-6
(49) Xiong, B.; Huang, Z.; Zou, H.; Qiao, C.; He, Y.; Yeung, E. S. ACS Nano 2017, 11, 541. doi: 10.1021/acsnano.6b06591
(50) Cao, X.; Feng, J.; Pan, Q.; Xiong, B.; He, Y.; Yeung, E. S. Anal.Chem. 2017, 89, 2692. doi: 10.1021/acs.analchem.6b03844
