Incoloy800H合金在高温纯水蒸气中的氧化行为
2018-01-19,,,,,
,, ,, ,
(西安热工研究院有限公司,电站锅炉煤清洁燃烧国家工程中心,西安 710032)
0 引 言
高温气冷堆(high temperature gas cooled reactor, HTGR)因其安全性高、系统简单、发电效率高且经济性好等特点,成为第四代核电站的首选堆型。在高温气冷堆中,一回路中的氦气冷却剂温度可达750~950 ℃,蒸汽发生器内壁以及U型传热管承受的平均温度为600 ℃(传热器过热段承受的温度则更高),U型管内外压力差达25 MPa,这对蒸汽发生器结构件包括U型传热管等部件用材料的高温性能提出了很高的要求[1]。Incoloy800H(Fe-32Ni-21Cr)合金是一种以奥氏体为基体组织的固溶强化型镍基高温合金,该合金通过在1 100~1 200 ℃进行固溶处理而获得稳定的奥氏体组织和所需的晶粒度,并使强化元素弥散分布在基体中以得到相应的强度。由于该合金还具有良好的耐高温和抗蠕变性能,因此被推荐用作高温气冷堆的蒸汽发生器等关键部件材料。
在超临界水环境中(温度高于374 ℃,压力高于22.1 MPa),氧化是合金主要的腐蚀方式。有关Incoloy800H合金在超临界水中的氧化行为已有不少报道[2-4]。OTSUKA等[4]研究发现,Incoloy800H合金在700 ℃纯水蒸气中形成了外层为Fe3O4、内层为(Fe,Cr)3O4的双层氧化膜。TAN等[5]研究了Incoloy800H合金在500 ℃/25 MPa超临界水中(溶解氧含量分别为2 μg·L-1和2.5×10-2μg·L-1)的氧化行为,得到了相似的研究结果,并进一步指出,在Fe3O4氧化层的外表面还形成一层连续的(Cr,Fe)2O3层。此外,TAN等[6-7]还发现在试验条件下氧化膜发生了严重剥落,通过晶界工程可显著改善氧化膜的抗剥落性能。然而,FULGER等[2]认为:在450~600 ℃/25 MPa的超临界水环境中,Incoloy800H合金表面形成了由外层NiFe2O4和内层FeCrO3组成的双层氧化膜;450 ℃时氧化膜的生长遵循立方规律,温度升高至600 ℃时氧化膜的生长动力学则遵循抛物线规律。CHEN等[8]对Incoloy800H合金在950 ℃湿空气中的氧化行为进行研究时发现:该合金的氧化动力学遵循抛物线规律,氧化膜由MCr2O4(此处M代表铁和铬)和Cr2O3组成,与内层的Cr2O3相比,外层MCr2O4较薄且疏松易剥落;在氧化膜下方的基体中,铝和硅分别内氧化形成Al2O3和SiO2。综上所述,Incoloy800H合金在水蒸气或含水蒸气环境中的氧化产物结构还不够明晰,关于该合金在纯水蒸气中氧化机制的研究仍较为匮乏。
为此,作者采用不连续称重法研究了Incoloy800H合金在750~850 ℃纯水蒸气中的氧化行为,探讨了该合金在高温纯水蒸气中的氧化机制。
1 试样制备与试验方法
1.1 试样制备
试验所用的Incoloy800H合金为供货态管材,其化学成分见表1。采用线切割制备出尺寸为15 mm×10 mm×1 mm的片状试样,用1200#SiC砂纸打磨后,置于丙酮中进行超声清洗,吹干备用。
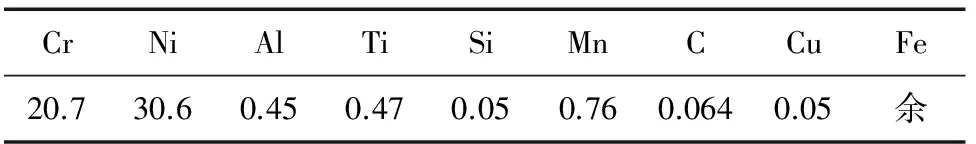
表1 Incoloy800H合金的化学成分(质量分数)Tab.1 Chemical composition of Incoloy800Halloy (mass) %
1.2 试验方法
在水平的管式马弗炉中,采用自主搭建的试验装置[9]进行蒸汽氧化试验。将试样悬挂在试样架上,置于石英反应管内,利用高纯N2先将石英反应管内的空气排除,待马弗炉加热至设定的蒸汽温度(750,850 ℃)时,关闭N2;将已预热至250 ℃的电阻率为18.25×108Ω·m的超纯水(溶解氧含量为5~6 mg·L-1)泵入到反应管内,生成的水蒸气流量为100~120 mL·s-1,温度分别为750,850 ℃,压力为1.01×105Pa,氧化时间为500 h。
采用不连续称重法测合金的氧化动力学曲线。将试样在水蒸气中氧化一定时间后,关闭水蒸气并通入N2,冷至室温,随后取出试样,干燥;使用精度为0.01 mg的赛多利斯电子天平称取质量,计算氧化前后的单位面积质量增加值Δm,然后将试样再次放入反应炉中继续氧化;循环此过程,得到不同氧化时间下的Δm,绘制氧化动力学曲线。
蒸汽氧化试验结束后,利用SHIMAZDU XRD-7000型X射线衍射仪(XRD)分析氧化产物的物相组成,采用铜靶,Kα射线,电压为40 kV,扫描速率为8 (°)·min-1,扫描角度范围为10°~100°;用ZEISS ∑IGMAHD型扫描电子显微镜(SEM)观察氧化膜的表面形貌和截面形貌,截面形貌观察时使用冷镶法制样,并使用附带的能谱仪(EDS)对氧化物进行成分分析。
2 试验结果与讨论
2.1 氧化动力学曲线
(Δm)2=kpt
(1)
式中:kp为氧化动力学抛物线速率常数。
由图1(b)可以得到试验合金在750,850 ℃水蒸气中氧化时的kp分别为1.61×10-10,1.13×10-8g2·cm-4·h-1。
2.2 氧化膜的物相组成
由图2(a)可知:在750 ℃纯水蒸气中氧化500 h后,试验合金表面的氧化物由(Fe,Mn)3O4和(Cr,Mn)2O3组成,(Cr,Mn)2O3的衍射峰强度明显较高,说明试验合金的表面氧化产物以(Cr, Mn)2O3为主;XRD谱中还出现了来自金属基体的衍射峰,且其强度远高于(Fe,Mn)3O4和(Cr,Mn)2O3的,说明此氧化膜很薄。由图2(b)可知,在850 ℃纯水蒸气中氧化500 h后,试验合金表面的氧化物组成与在750 ℃纯水蒸气中氧化后的相同,但(Fe,Mn)3O4和(Cr,Mn)2O3的衍射峰强度相对增强,而基体的衍射峰强度减弱,说明氧化膜的厚度增大。
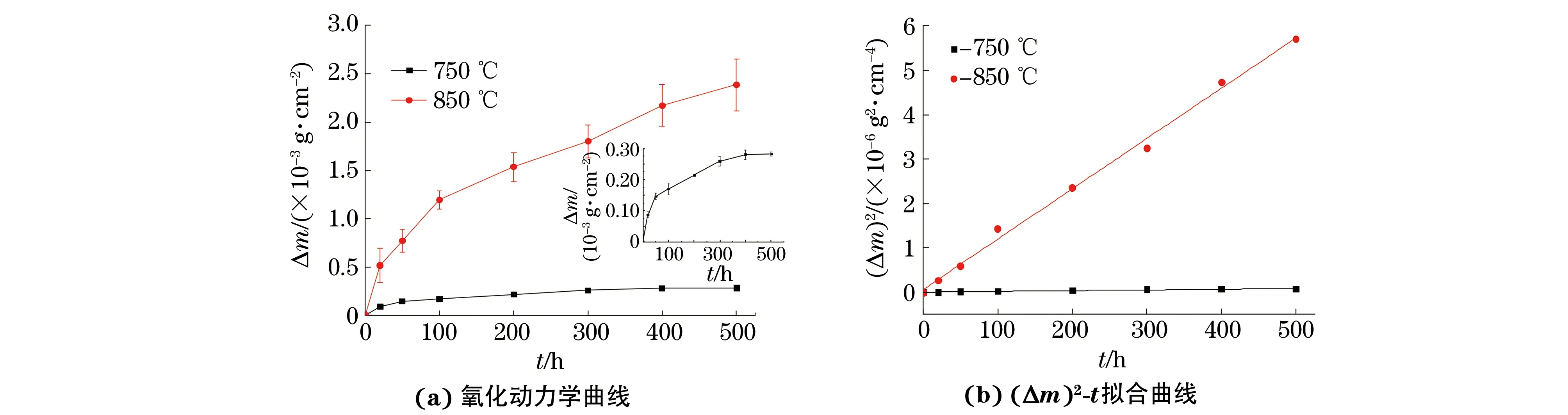
图1 在不同温度纯水蒸气中氧化时试验合金的氧化动力学曲线和(Δm)2-t拟合曲线Fig.1 Oxidation kinetic curves (a) and (Δm)2-t fitting curves (b) of tested alloy during oxidation in pure steam at different temperatures

图2 在不同温度纯水蒸汽中氧化500 h后试验合金表面的XRD谱Fig.2 Surface XRD patterns of tested alloy after oxidation in pure steam for 500 h at different temperatures

图3 在不同温度纯水蒸气中氧化500 h后试验合金的表面和截面形貌Fig.3 Surface (a,c) and cross-section morphology (b,d) of tested alloy after oxidation in pure steam at different temperatures for 500 h
2.3 氧化膜的微观形貌
由图3(a)、图3(b)可知:在750 ℃纯水蒸气中氧化500 h后,试验合金表面形成凹凸不平的氧化膜,突起处的氧化膜较为疏松,平整区域的则较为致密;放大后可观察到平整区域被片状氧化物覆盖,而突起区域由尺寸较大的颗粒状氧化物组成;氧化膜与基体的界面较为平整,突起处的氧化膜分为两层,外层的衬度为灰色而内层的为深灰色,且外层氧化膜的厚度远大于内层氧化膜的,平整区域的衬度为深灰色,氧化膜为单层结构。EDS分析表明,片状氧化物富铬,颗粒状氧化物富铁。结合图2(a)分析可知,这些氧化物分别由Cr2O3和Fe3O4组成。片状Cr2O3的形成与离子沿缺陷如晶界、位错等“短路扩散通道”的快速扩散有关[10-12]。
由图3(c)、图3(d)可知:当纯水蒸气的温度升至850 ℃时,试验合金表面氧化膜的形貌衬度差异消失,未出现片状和突起状的氧化物;与750 ℃的相比,在850 ℃纯水蒸气中氧化后,氧化物的晶粒尺寸增大,氧化膜的厚度增加且更为均匀,平均厚度为(19.2±2.9)μm,与图1的氧化动力学结果一致;氧化膜呈外层疏松、内层致密的双层结构,如图3(d)中箭头所指,且外层氧化膜的厚度远小于内层氧化膜的;在氧化膜下方的基体中沿晶界发生了内氧化。
由图4可知:在750 ℃纯水蒸气中氧化500 h后,试验合金表面突起处的外层氧化膜富铁,内层氧化膜富铬并含有少量的锰。由此判断,外层氧化膜和内层氧化膜的组成分别为Fe3O4和(Cr, Mn)2O3。结合图3(b)分析还可发现,在氧化膜下方的基体中形成了富氧区,且氧的面分布图与铬的重合,这说明此区域为铬的内氧化区。结合图2(a)分析可知,此内氧化产物为Cr2O3,在内氧化区前沿,Cr2O3近乎连续。

图4 在750 ℃纯水蒸气中氧化500 h后试验合金的截面形貌及元素面扫描结果Fig.4 Cross-section morphology (a) and elemental mapping results (b-h) of tested alloy after oxidation in pure steam at 750 ℃ for 500 h

图5 在850 ℃纯水蒸气中氧化500 h后试验合金的截面形貌及元素面扫描结果Fig.5 Cross-section morphology (a) and elemental mapping results (b) of tested alloy after oxidation in pure steam at 850 ℃ for 500 h
由图5可以看出:在850 ℃纯水蒸气中氧化500 h后,试验合金表面外层氧化膜富铁和锰,而内层氧化膜富铬和锰,结合图2(b)分析可知,内、外层氧化膜分别由(Cr, Mn)2O3和(Fe, Mn)3O4组成;氧化膜下方基体的晶界处发生铝富集,且其面分布图与氧的重叠,这表明铝发生了内氧化,说明图3(d)中的晶界内氧化产物为Al2O3。在对湿空气中Incoloy800H合金的氧化行为进行研究时,CHEN等[8]有同样的发现,且认为铝的内氧化产物为Al2O3。
2.4 氧化机制
综上所述:Incoloy800H合金在750 ℃纯水蒸气中氧化后形成连续的(Cr,Mn)2O3膜,在此膜外表面形成了不连续、突起状的Fe3O4,在氧化膜下方的基体中铬发生内氧化形成Cr2O3;在850 ℃纯水蒸气中氧化后则形成双层氧化膜,其中外层为薄的(Fe, Mn)3O4膜,内层为厚的(Cr,Mn)2O3膜,氧化膜下的基体中铝发生内氧化形成Al2O3。在此基础上对Incoloy800H合金在高温纯水蒸气中的氧化机制进行探讨。

图6 在750 ℃纯水蒸气中氧化50 h后试验合金的表面形貌Fig.6 Surface morphology of tested alloy after oxidation in pure steam for 50 h at 750℃
根据经典氧化理论,在氧化初期,合金表面铁、铬的氧化物Fe3O4和Cr2O3同时形核,见图6。与菱方结构的Cr2O3相比,立方结构Fe3O4中的缺陷浓度较高,这使得后者以较快的速度生长[13-15]。Fe3O4的生长是不连续的,往往止于晶界,如图3(b)中箭头所指。其原因可能在于,Cr3+、O2-沿晶界的扩散速率比在晶内的快,富铬氧化物易在晶界处形成,从而抑制了晶界处富铁氧化物的形成[16-17]。Cr3+的扩散促使Cr2O3沿横向和纵向两个方向生长,最终形成连续的Cr2O3膜。一旦连续、致密的Cr2O3膜形成,合金的氧化质量增加速率就会降低。这是因为Cr2O3是一种偏离化学计量比极小的氧化物,其晶格中的缺陷浓度极低,因而阴、阳离子通过Cr2O3的扩散速率很低[18-20]。在Cr2O3的生长过程中,由于Mn3+在Cr2O3中的扩散速率远大于Cr3+在其中的扩散速率,Mn3+源源不断地扩散进入Cr2O3的晶格内,形成(Cr, Mn)2O3固溶体[21]。综上,氧化膜的生长受阴、阳离子通过氧化膜的扩散控制,因此合金的氧化动力学遵循抛物线规律[22-23]。
当水蒸气温度升高后,合金的氧化行为发生变化:①离子(和原子)在氧化物(和基体金属)中的扩散加速,合金的氧化速率增加,氧化膜增厚;②(Cr, Mn)2O3的快速生长一方面使得Fe2+/Fe3+的扩散距离增大,另一方面铬可将已经氧化的铁还原出来,这降低了外层Fe3O4的生长速率,导致其厚度远小于内层(Cr, Mn)2O3的;③阴离子(O2-)、阳离子(Cr3+)在氧化物晶格内及晶界处的扩散速率均增大,在晶格内的扩散变得与在晶界处的同等重要[24],(Cr,Mn)2O3在晶界的择优生长不再明显,因而对Fe3O4在晶界生长的抑制作用减弱,Fe3O4生长成连续的一层;④锰可通过内层(Cr,Mn)2O3扩散至Fe3O4从而形成(Fe,Mn)3O4固溶体。与在750 ℃水蒸气中氧化时的相比,在850 ℃水蒸气中氧化时(Cr, Mn)2O3的生长大幅加速,铬的消耗增大,氧化膜下方基体中铬含量进一步降低(质量分数约8%,而750 ℃时与氧化膜毗邻基体中铬质量分数约为14%)。根据内氧化产物的形核机制模型[25-28],内氧化区前沿处新的内氧化产物(Cr2O3)晶核的形成必须满足一定的条件,即
(2)
式中:Ksp为内氧化产物的浓度积;NO(s)为氧化膜与基体界面处氧的含量;NB(O)为合金中溶质元素(铬)的含量。
由此推测:试验合金在850 ℃水蒸气中氧化时未形成Cr2O3内氧化物的原因在于其基体中铬的质量分数过低,不满足Cr2O3形核所需的临界过饱和度;而由于铬含量较少,基体中铝的相对含量增加,化学稳定性更高的Al2O3形核成为可能;随着深度的增加,铝的内氧化程度降低。
3 结 论
(1) 在750 ℃和850 ℃纯水蒸气中氧化时,Incoloy800H合金的氧化动力学遵循抛物线规律,水蒸气温度升高后合金的氧化速率增大。
(2) 在750 ℃纯水蒸气中氧化后,Incoloy800H合金表面形成连续致密的(Cr, Mn)2O3膜,其表面存在不连续、突起状且结构疏松的Fe3O4层;纯水蒸气温度升高至850 ℃后,Fe3O4生长成为连续的一层且其中有少量锰富集,其厚度远小于内层(Cr, Mn)2O3膜的厚度。
(3) 在750 ℃纯水蒸气中氧化后,表面氧化膜中(Cr, Mn)2O3层下方基体中的铬被内氧化生成Cr2O3;温度升高至850 ℃,在氧化膜下方的基体中仅有铝沿晶界发生内氧化形成Al2O3。
[1] 尹清辽,孙玉良,居怀明,等. 模块式高温气冷堆超临界蒸汽发生器设计[J]. 原子能科学技术,2006,40(6):707-713.
[2] FULGER M, OHAI D, MIHALACHE M,etal. Oxidation behavior of Incoloy 800 under simulated supercritical water conditions [J]. Journal of Nuclear Materials, 2009, 385(2): 288-293.
[3] TAN L, ALLEN T R, YANG Y. Corrosion behavior of alloy 800H (Fe-21Cr-32Ni) in supercritical water [J]. Corrosion Science, 2011, 53(2): 703-711.
[4] OTSUKA N, FUJIKAWA H. Scaling of austenitic stainless steels and nickel-base alloys in high-temperature steam at 973 K[J]. Corrosion, 1991, 47(4): 240-248.
[5] TAN L, REN X, SRIDHARAN K,etal. Effect of shot-peening on the oxidation of alloy 800H exposed to supercritical water and cyclic oxidation [J]. Corrosion Science, 2008, 50(7): 2040-2046.
[6] TAN L, SRIDHARAN K, ALLEN T R. The effect of grain boundary engineering on the oxidation behavior of INCOLOY alloy 800H in supercritical water [J]. Journal of Nuclear Materials, 2006, 348(3): 263-271.
[7] TAN L,SRIDHARAN K,ALLEN T R,etal.Microstructure tailoring for property improvements by grain boundary engineering [J]. Journal of Nuclear Materials, 2008, 374(1/2): 270-280.
[8] CHEN W S, KAI W, TSAY L W,etal. The oxidation behavior of three different zones of welded Incoloy 800H alloy [J]. Nuclear Engineering and Design, 2014, 272: 92-98.
[9] 杨珍,鲁金涛,赵新宝,等. HR3C在750 ℃空气和水蒸气中的高温氧化行为研究[J]. 动力工程学报,2015,35(10):859-864.
[10] TALLMAN R L, GULBRANSEN E A. Dislocation and grain boundary diffusion in the growth of α-Fe2O3whiskers and twinned platelets peculiar to gaseous oxidation [J]. Nature, 1968, 218: 1046-1047.
[11] VOSS D A, BUTLER E P, MITCHELL T E. The growth of hematite blades during the high temperature oxidation of iron [J]. Metallurgical Transactions A, 1982, 13(5): 929-935.
[12] RAYNAUD G, RAPP R. In situ observation of whiskers, pyramids and pits during the high-temperature oxidation of metals [J]. Oxidation of Metals, 1984, 21(1/2): 89-102.
[13] LIMOGE Y, BOCQUET J L. Selfdiffusion and point defects in iron oxides FeO, Fe3O4, α-Fe2O3[J]. Defect and Diffusion Forum, 2001, 194: 1051-1056.
[14] HAGEL W C, SEYBOLT A U. Cation diffusion in Cr2O3[J]. Journal of the Electrochemical Society, 1961, 108(12): 1146-1152.
[15] HAGEL W C. Anion diffusion in α-Cr2O3[J]. Journal of the American Ceramic Society, 1965, 48(2): 70-75.
[16] JUTTE R H, KOOI B J, SOMERS M A,etal. On the oxidation of α-Fe and ε-Fe2N1-z: I. Oxidation kinetics and microstructural evolution of the oxide and nitride layers [J]. Oxidation of Metals, 1997, 48(1/2): 87-109.
[17] STOTT F, WEI F. High temperature oxidation of commercial austenitic stainless steels [J]. Materials Science and Technology, 1989, 5(11): 1140-1147.
[18] KOFSTAD P K. Nonstoichiometry, diffusion and electrical conductivity in binary metal oxides [M]. New York: Wiley-Interscience, 1972.
[19] KOFSTAD P, LILLERUD K. Chromium transport through Cr2O3scales I. On lattice diffusion of chromium [J]. Oxidation of Metals, 1982, 17(3/4): 177-194.
[20] GRESKOVICH C. Deviation from stoichiometry in Cr2O3at high oxygen partial pressures [J]. Journal of the American Ceramic Society, 1984, 67(6): C111-C112.
[21] LOBNIG R E, SCHMIDT H P, HENNESEN K,etal. Diffusion of cations in chromia layers grown on iron-base alloys [J]. Oxidation of Metals, 1992, 37(1/2): 81-93.
[22] WAGNER C. Reaktionstypen bei der oxydation von legierungen[J].Berichte der Bunsengesellschaft für Physikalische Chemie, 1959, 63(7): 772-782.
[23] WAGNER C. Theoretical analysis of the diffusion processes determining the oxidation rate of alloys [J]. Journal of the Electrochemical Society, 1952, 99(10): 369-380.
[24] SUZUOKA T. Lattice and grain boundary diffusion in polycrystals [J]. Transactions of Japan Institute of Metals, 1961, 2: 25-33.
[25] LAFLAMME G R,MORRAL J E. Limiting cases of subscale formation[J].Acta Metallurgica, 1978, 26(12): 1791-1794.
[26] KAMPMANN L, KAHLWEIT M. Zur theorie von fällungen [J]. Berichte der Bunsengesellschaft für Physikalische Chemie, 1970, 74(5): 456-462.
[27] WAGNER C. Mathematical analysis of the formation of periodic precipitations [J]. Journal of Colloid Science, 1950, 51(1): 85-97.
[28] YALI L, MORRAL J E. A local equilibrium model for internal oxidation[J].Acta Materialia,2002,50(14):3683-3691.
