气相色谱-质谱法测定十种植物生长调节剂残留
2018-01-12刘环宇郭志东
刘环宇 郭志东
(广东药科大学医药化工学院,广东 中山 528458)
目前农药残留的检测已由单一农药品种检测发展为多农药残留组分同时检测,而色谱质谱联用技术在这方面的应用最多[1~4]。采用超临界流体萃取(SFE)、柱前衍生GC-MS测定果蔬中2,4-D等四种农药,前处理过程无需净化,经2,2,2-三氯乙醇或溴甲基五氟苯衍生化后直接进行分析,与传统的溶剂提取相比更加经济、快速,且回收率、检测限和重现性都符合残留检测的要求。采用液相色谱-大气压化学电离-离子阱-三重四极杆串联质谱分析了柑橘中抑霉唑等六种农药,样品经乙酸乙酯提取,在单级、串联和三级质谱模式下的最低检出限为0.000 5~0.3 mg/kg,回收率72~94%,RSD<19%。虽然三级质谱的灵敏度低于单级和串联质谱,但选择性较好[5~8]。吴凤琪[9]、王静静[10]等分别建立了水果中八种外源性调节剂(乙烯利、丁酰肼、抑芽丹、赤霉素、玉米素、氯吡脲、矮壮素、β-萘乙酸)的SPE-LC-MS/MS技术和果蔬中六种植物生长抑制剂(氯化胆碱、矮壮素、缩节胺、嘧啶醇、多效唑、烯效唑)的SPE-HPLC-ESI-MS/MS技术。
色谱质谱联用技术可以简化前处理程序、缩短检测时间、减少误判、提高准确度,是国际公认具有法律效力的标准方法。常用的色谱法依靠保留时间定性,假阳性和假阴性是不可避免的,为了提高检测结果的可靠性,色谱质谱联用技术(特别是串联质谱技术)的推广是必然的。
1 仪器与设备
1.1 仪器
气相色谱-串联质谱仪;氮吹仪;固相萃取仪;TG-SMS毛细管色谱柱(30 m×0.25 mm×0.25 μm)
1.2 试剂
酸性橙II标准品;酸性金黄标准品;碱性嫩黄O标准品;提取溶液:乙腈;洗脱溶液:甲醇-50mmol/L乙酸铵。乙腈(色谱纯);甲醇(色谱纯);乙酸乙酯(色谱纯);正己烷(色谱纯);三氟化硼乙醚溶液(分析纯);甲酸(分析纯);盐酸(分析纯);氯化钠(分析纯);无水硫酸钠(分析纯);QuECHERS试剂盒(300 mg/管);MCS固相萃取柱(500mg/6mL);2,4-D-乙酯标准品;2,4-D-丁酯标准品;4-氯苯氧乙酸(CPA)标准品;2,4-二氯苯氧乙酸标准品;β-萘乙酸标准品;吲哚乙酸标准品;吲哚丁酸标准品;多效唑标准品;激动素标准品;6-苄基腺嘌呤标准品。
1.3 标准溶液及试剂配制
1.0 g/L植物生长调节剂(十种)混合贮备液:分别称取0.025 g上述植物生长调节剂标准品,用甲醇分别溶解并定容至25mL,并在4 ℃保存。5.0 mg/L植物生长调节剂混合应用液:各吸取1.0 g/L植物生长调节剂贮备液0.5 mL,用甲醇定容至100 mL,并在4 ℃保存。5%氨化甲醇:取5 mL氨水,用甲醇定容到100 mL,并且现配现用。20%乙酸乙酯-正己烷混合液:吸取2 mL乙酸乙酯和8 mL正己烷混合而成。10%三氟化硼甲醇衍生溶液:吸取1 mL三氟化硼乙醚溶液和9 mL甲醇混合而成,并且现配现用。
1.4 样品的提取及分级净化
1.4.1生长调节剂提取
称取捣碎的豆芽试样10.000 g于50 mL离心管中,加入20 mL乙腈,40 μL甲酸,漩涡混合均匀大约1 min,超声提取约30 min,转速8 000 r/min状态下离心5 min,上清液转移至另一支50 mL离心管中,加入3.0 g NaCl,漩涡混合均匀;8 000 r/min转速下离心5 min,然后吸出乙腈层,用无水Na2SO4脱水后收集至圆底烧瓶内,然后用50 ℃水浴真空浓缩至干,最后在圆底烧瓶内加入2 mL甲醇进行超声溶解。
1.4.2生长调节剂净化
取上述甲醇样液1 mL,加到QuECHERS试剂管中,混合均匀,静止5 min混合均匀,10 000 r/min转速下离心2 min,取上清液进行GC/MS分析测定2,4-D-乙酯和2,4-D-丁酯。另取甲醇样液1 mL,加入9 mL 40 mmol/L HCl,超声混合均匀,转移至离心管后在8 000 r/min转速下离心5 min,上清液等待净化。然后先用5 mL甲醇、5 mL水、5 mL 40 mmol/L HCl溶液来活化MCS柱,活化结束后把上清液转移到MCS柱内,待样液过柱后,用5 mL水淋洗除杂,真空抽干柱内液体后加入5 mL甲醇洗脱,收集于10 mL具塞试管内,得组分Ⅰ;用5 mL 5%氨化甲醇洗脱,收集于10 mL具塞试管内,得组分Ⅱ,洗脱液分别于50 ℃下用氮气吹干。组分Ⅱ用0.5 mL甲醇溶解后进行GC/MS分析,测定多效唑、激动素、6-BA。
1.5 衍生化
组分Ⅰ加入1 mL 10%三氟化硼甲醇衍生溶液,涡旋混合均匀,70 ℃加热衍生30 min,取出冷却后再加入1.0 mL 20%乙酸乙酯/正己烷混合液和2 mL纯水,涡旋混合均匀,4 000 r/min转速下离心5 min,吸出上层有机相,转移至进样瓶中进行GC/MS分析。分别吸取5.0 mg/L植物生长调节剂混合应用液0.05,0.10,0.20,0.40和1.00 mL,以甲醇定容至1.0 mL,与样品一起进行衍生测定。
1.6 色谱和质谱条件
TG-SMS毛细管色谱柱(30 m×0.25 mm×0.25 μm);进样口温度:260 ℃;柱温:初温80 ℃,保持1 min,以10 ℃/min升至300 ℃,300 ℃后保留2 min;载气:氦气,纯度≥99 %,流速1 mL/min;进样量:1 μL;电离方式:EI源,70eV;离子源温度:230 ℃;进样方式:不分流进样;监测方式:扫描范围m/z:35~450。
2 结果与讨论
2.1 生长调节剂提取
豆芽中植物生长调节剂多残留测定提取溶剂主要有甲醇、乙腈,考虑乙腈提取可与目前国家标准方法提取溶剂相一致,优先选择乙腈作为提取溶剂。研究中的五种植物生长调节剂含羧基,为了提高提取率,在乙腈中适当添加甲酸可以明显改善植物生长调节剂的提取效率。实验表明,不加甲酸,十种植物生长调节剂平均回收率为64.6~92.3%(n=3),添加甲酸的乙腈可以增加带羧基植物生长调节剂的提取率,十种植物生长调节剂回收率在83.2~94.2%之间(n=3)满足豆芽中植物生长调节剂提取要求。
2.2 样品分级净化
十种植物生长调节剂按照其化学结构式,在酸性条件下可以分成两类,一类中性,如2,4-D-乙酯、2,4-D-丁酯、CPA、β-萘乙酸、2,4-D、吲哚乙酸、吲哚丁酸;另一类碱性带正离子,如多效唑、激动素、6-BA。中性化合物中2,4-D-乙酯,2,4-D-丁酯,可以直接用GC/MS测定。CPA,β-萘乙酸、2,4-D、吲哚乙酸和吲哚丁酸五种植物生长调节剂需要衍生后测定。采用三氟化硼甲醇甲酯化衍生,改善了峰形、提高了检测灵敏度;碱性化合物多效唑、激动素、6-BA可以直接进行GC/MS分析。
2.2.1QuECHERS净化
QuEChERS技术具有试剂用量少、快速等优点,克服了凝胶色谱和固相萃取试剂用量大的缺点,已经广泛应用于植物样品中农药残留的测定。豆芽中干扰物质主要有色素、脂肪及脂肪酸、嘌呤等,选择含C18、石墨化碳黑和PSA填料的试剂盒,可以除去豆芽提取物中色素、脂肪酸等,2,4-D-乙酯,2,4-D-丁酯不被填料吸附,豆芽加标测定图谱(见图1)。
2.2.2固相萃取分级净化
牟艳利等[11]采用MAX混合型阴离子固相萃取柱结合LC/MS净化测定瓜果中7种酸性植物生长调节剂。由于多种植物生长调节剂测定难在同一气相色谱条件下完成测定,先将不同性质的植物生长调节剂分离后分别测定。通过豆芽加标回收实验,比较了MCX,PCX,SLW,MCS等混合型阳离子固相萃取柱对上述八种植物生长调节剂分级净化效果。实验结果表明,MCX,PCX,SLW对吲哚乙酸、吲哚丁酸回收率较差;而MCS柱对八种植物生长调节剂回收率均在80%以上,且除杂效果较好。用甲醇从MCS柱洗脱获得组分Ⅰ,组分Ⅰ中有CPA,β-萘乙酸、2,4-D-吲哚乙酸、吲哚丁酸等含羧基的五种植物生长调节剂,甲酯化后进行GC/MS分析,样品加标总离子流图(见图2);5%氨化甲醇从MCS柱洗脱组分Ⅱ,组分Ⅱ含多效唑、激动素、6-BA这三种植物生长调节剂,这三种组分可以直接进行GC/MS分析,样品加标总离子流图(见图3)。
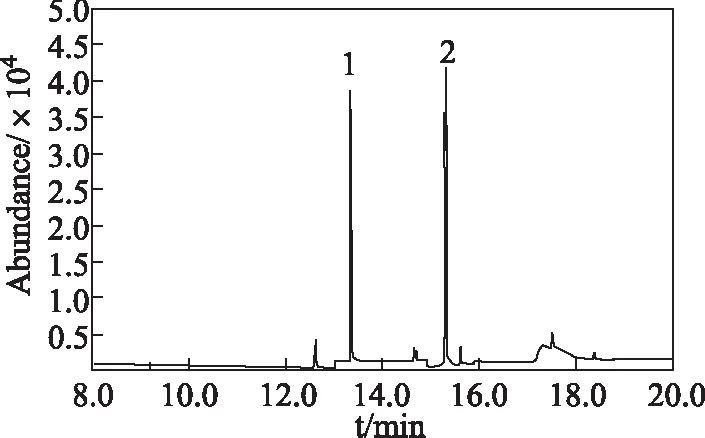
图1 豆芽加标2,4-D-乙酯、2,4-D-丁酯测定总离子流Fig 1 Total ion chromatogram of bean sproutspiking 2,4-D-ethyl ester and 2,4-D-butyl ester

图2 组分Ⅰ甲酯化后测定总离子流图Fig 2 Total ion chromatogram of compositionⅠafter methyl esterification
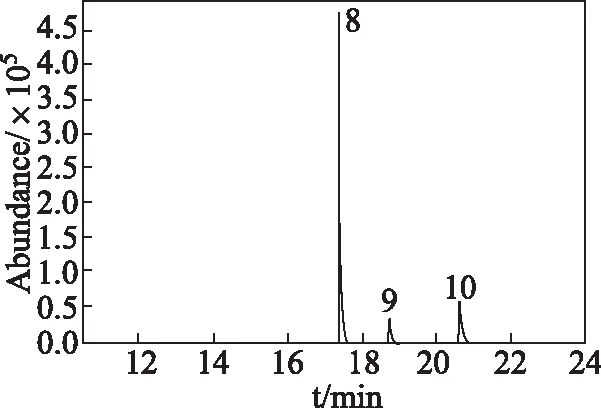
图3 组分Ⅱ测定总离子流图Fig 3 Total ion chromatogram of compositionⅡ
由图2和图3可知,通过MCS柱对豆芽中八种植物生长调节剂进行分级净化,采用甲醇和5%氨化甲醇分别洗脱不同的植物生长调节剂,分别进行GC/MS分析,就可有效除去干扰物质,使得实验材料纯度比较高,可以满足实验要求。
2.3 气相色谱质谱条件
目前,植物生长调节剂的多组分测定大多数采用LC/MS/MS法,虽然其定性准确、灵敏度高,适用极性高的物质测定,但定量分析的基质效应十分明显,同样需要对样品进行合理的净化,如进行QuECHERS净化、固相萃取净化,另一方面,LC/MS/MS法仪器成本高,方法推广性差。GC/MS对极性较高的物质需要进行衍生化等措施来改善化合物的性质,仪器相对便宜且较为普及,采用GC/MS来评价豆芽中十种植物生长调节剂分级净化效果。在当前色谱条件下,采用TG-SMS柱,豆芽中十种植物生长调节剂均能较好分离,各化合物GC/MS分析条件(见表1)。十种植物生长调节剂在0.025~1.0 mg/L浓度范围内线性较好,相关系数约为0.995~0.999最低检测浓度为0.02~0.05 mg/L。
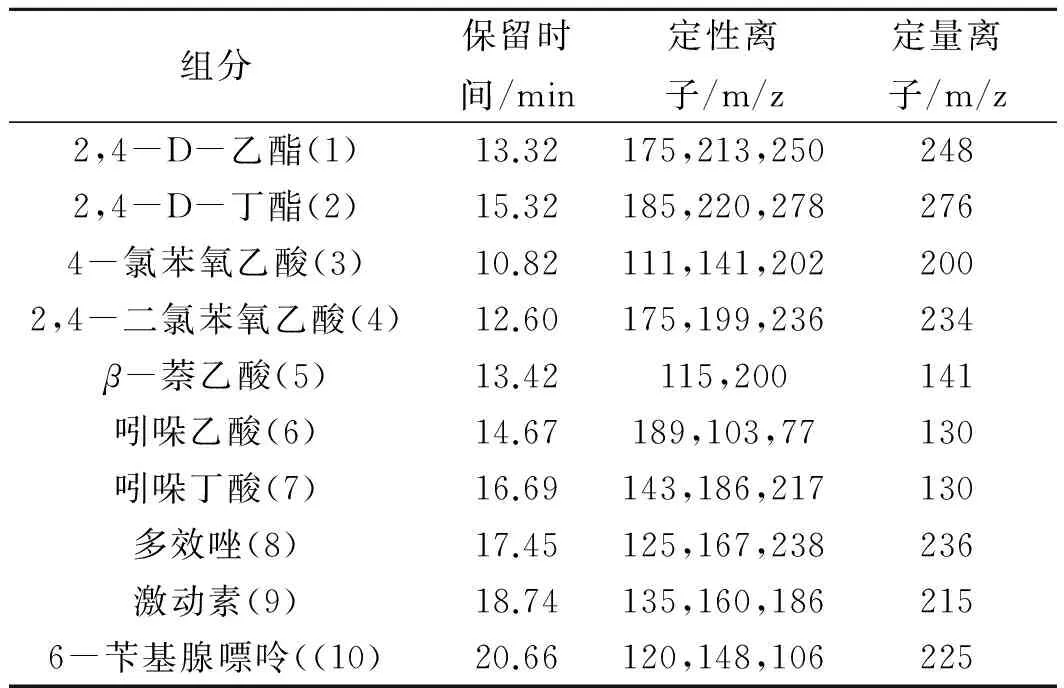
表1 十种植物生长调节剂的保留时间与监测离子Table 1 Monitoring ions,rententiontimes for ten plant growth regulators

表2 方法加标回收率、检出限、定量限和精密度Table 2 Recoveries,determination limits,quantification limits and precisions of method
2.4 方法精密度、检出限及样品测定
根据本实验所建立的方法,在豆芽中进行加标回收实验,结果(见表2)。由表2可知,十种植物生长调节剂在豆芽中添加0.01~0.10 mg/kg,平均回收率为70.5~93.2%,RSD为5.2~12.3%,本方法对十种植物生长调节剂的定量限为0.010~0.025 mg/kg,根据3倍信噪比计算,检出限为0.003~0.008 mg/kg。
3 结 论
建立了分级净化结合气相色谱-质谱法测定植物中2,4-D-乙酯、2,4-D-丁酯、CPA、β-萘乙酸、2,4-D、吲哚乙酸、吲哚丁酸、多效唑、激动素、6-BA这十种植物生长调节剂残留的分析检验方法。结果表明,本方法完全可以用于豆芽中十种植物生长调节剂残留的检测,在豆芽中添加0.01~0.10 mg/kg十种植物生长调节剂平均回收率范围为70.5~93.2%,RSD为5.2~12.3%,本方法对十种植物生长调节剂的定量限(S/N,10)为0.0100~0.025 mg/kg,检出限(S/N,3)为0.003~0.008 mg/kg。此净化体系具有简便、快速、准确等优点,结合GC/MS可以满足豆芽中植物生长调节剂多残留的检测要求。
[1] 《食品中农业化学品残留限量》编委会. 食品中农业化学品残留限量(药品卷)[M]. 北京: 中国标准出版社, 2006.
[2] 中华人民共和国卫生部. GB2763-2005 食品中农药最大残留限量[M]. 北京: 中国标准出版社, 2005.
[3] 刘环宇,杨剑峰,吕君亮,王春丽. 高效液相色谱-串联质谱法同时测定食品中非法添加的三种偶氮类工业染料[J]. 化工时刊, 2014, 28(12): 15~17,58.
[4] 周艳明,忻雪. 高效液相色谱法测定果蔬中7种植物激素的残留量[J]. 食品科学, 2010, 31(18): 301~304.
[5] 储晓刚,雍炜,蔡慧霞 等. 顶空气相色谱法快速测定浓缩菠萝汁中乙烯利的残留量[J]. 色谱, 2001, 19(3): 286~288.
[6] 李丽华, 郑玲. 固相微萃取-气相色谱联用技术测定芒果原浆中乙烯利的残留量[J]. 分析试验室, 2007(增刊1): 287~289.
[7] 周艳明, 韩瑜, 田宏哲 等. 高效液相色谱-质谱法对水果中矮壮素的检测[J]. 分析测试学报, 2009, 28(10): 1 206~1 908.
[8] 周艳明, 韩瑜, 田宏哲 等. 高效液相色谱-质谱法测定蔬菜中矮壮素残留[J]. 食品科学, 2010, 31(14): 197~200.
[9] 吴凤琪, 靳保辉, 陈波 等. 水果中8种外源性植物生长调节剂的液相色谱-串联质谱测定[J]. 中国农学通报, 2010, 26(15): 115~119.
[10] 王静静, 鹿毅, 杨涛 等. HPLC-MS/MS法同时测定果蔬中6种植物生长抑制剂残留[J]. 分析测试学报, 2011, 30(2): 128~134.
[11] 牟艳莉, 郭德华, 丁卓平. 瓜果中常用植物生长调节剂的限量及检测方法[J]. 农药, 2013, 52(6): 398~401.
