心肌病与遗传△
2018-01-06张力力王树水
张力力 ,王树水
[1.南方医科大学,广州510100;2.广东省心血管病研究所心儿科广东省人民医院(广东省医学科学院),广州 510100]
心肌病与遗传△
张力力1,2,王树水1,2
[1.南方医科大学,广州510100;2.广东省心血管病研究所心儿科广东省人民医院(广东省医学科学院),广州 510100]
随着基因测序技术的飞速发展,心肌病遗传学研究方面取得了很大进展,已经发现了相关的多个致病基因和突变,并在发病机制方面进行了探索。但基因型和临床表型的关系仍不完善,基因突变和疾病的发生和预后之间的关系仍是尚未解决的难题。基因筛查作为遗传性疾病诊断的重要手段,在协助诊断心肌病,发现亚临床患者,筛查先证者的家属,指导治疗和预防方面有重要的作用,但仍面临着很多问题。现对心肌病的临床特点、发病机制、遗传学的研究进展进行综述。
心肌病;遗传学研究;基因;突变
1 心肌病的分类
目前,对于心肌病与遗传学方面的认知有了很大的进展,美国心脏协会(AHA)和美国心脏病学会(ESC)在最近这30年更新了心肌病的定义和分类。1980年世界卫生组织的分类,心肌病归为“不明原因的心肌病变”。1995年世界卫生组织第二次分类,提出“与心功能障碍相关的心肌疾病”,以及第一次包括了致心律失常型右心室心肌病/发育不良(arrhythmogenic right ventricular cardiomyopathy/dysplasia,ARVC/D)以及原发性限制型心肌病(restrictive cardiomyopathy,RCM)。2006年美国心脏协会发表科学文件提议,心肌病是表现为机械和电活动障碍的一组异质性心肌疾病,通常会出现(非必须)异常的心室肥厚或者扩张,这些形态学差异与不同的致病基因突变相关,第一次尝试根据基因进行心肌病的分类[1]。2008年欧洲心脏病学会(ESC)科学声明专家组提议,心肌病是心脏结构和功能障碍的心肌疾病,强调疾病的表型是治疗疾病的基础,根据形态功能学将心肌病分为扩张型心肌病(dilated cardiomyopathy,DCM)、肥厚型心肌病(hypertrophic cardiomyopathy,HCM)、RCM、AVRC和未命名的心肌病。这些类型又进一步分为家族遗传性和非家族遗传性两种类型[2]。
2013年世界心脏联盟(WHF)建议心肌病的基因型-表型MOGE(S)分类,即对心肌病从5个方面进行分类,包括功能特性(M),累及的器官(O),遗传模式(G),明确的病因(包括详细的遗传学缺陷或其他疾病原因)(E),按照美国心脏病学会/美国心脏协会(ACC/AHA)分级(A-D)和纽约心脏协会(NYHA)心功能Ⅰ~Ⅳ级进行功能状态分级(S)[3]。MOGE(S)分类同时从临床和遗传学方面对疾病进行最全面的描述,具有多种优势。专家期待这种命名方式进一步修改、完善。使我们在心肌病的诊断和治疗中,对疾病有更深刻的理解,有利于医生之间交流,促进多中心、多名族的注册研究,提高心肌病的诊断与治疗水平[3]。
1.1 扩张型心肌病
DCM是以心腔扩大,收缩功能减退为主要特征的心肌疾病,是心血管疾病中导致死亡和心脏移植的主要疾病,其组织学特点包括心肌坏死和纤维化,临床常表现从无症状到充血性心力衰竭、心律失常、血栓栓塞甚至猝死,约70%~90%的患儿在确诊时以心力衰竭为主要表现。为早期防治DCM,2016年欧洲心脏学会(ESC)心肌心包疾病专家组新增了收缩功能减低非DCM(HNDC)的类型[4]。2007年美国报道DCM的发病率为1/2 500,男性多于女性,DCM年发病率约为0.58/100 000,在各类型心肌病中约占50%[5]。引起心脏呈DCM改变的病因有多种,既有遗传因素又有非遗传因素,可以为一些全身性疾病,如感染性疾病(心肌炎16%)、代谢性疾病、内分泌疾病、结缔组织疾病、神经肌肉病(9%)、过敏性反应及毒素作用、先天畸形等,也可以为特发性、家族性或遗传性等[6]。家族性DCM定义为DCM患者家族中有两个或以上家族成员患有特发性DCM(IDCM),家族性占所有DCM的35%~48%。DCM的遗传模式包括常染色体显性遗传、染色体隐形遗传、X连锁和线粒体遗传等方式,主要以常染色体显性和隐性遗传为主。
到目前为止,已经发现37个基因相关位点的突变能致DCM,主要编码肌小节相关蛋白(ACTC1,ACTN2,CSRP3,MYBPC3,MYH6,MYH7,MYPN,TCAP,TNNC1,TNNI3,TNNT2,TPM1,TTN);细胞骨架相关蛋白(DSE,DMD,ILK,LAMA4,LDB3,PDLIM3,SGCD,VCL)核纤层蛋白(LMNA,TMPO)等[7]。近来有文献报道,与ARVC相关的编码桥粒蛋白(DSC2,DSG2,DSP)及一些离子通道基因(ABCC9,SCN5A)也与DCM相关[8-10]。其他与DCM有关的基因也编码肌浆网(PLN),剪接体(RBM20),线粒体(TAZ),γ分泌酶活动(PSEN1,PSEN2)等[7]。DCM最常见的基因突变是TTN基因,其在家族性和散发性患者的突变频率分别是25%和18%[11]。许多基因突变通过不同的病理、生理机制可引起DCM,但由于遗传异质型与临床表型异质性、遗传基因外显不全并呈年龄依赖性、遗传方式多样性,家族性DCM的识别和诊断具有一定的局限性。
1.2 肥厚型心肌病
HCM是以不能解释的心室肥厚为特征的心肌疾病,非对称性肥厚表现为室间隔与左心室游离壁之比>1.3,其他表现包括对称性、心尖肥厚,早期伴心脏舒张功能不全,晚期收缩功能亦受影响,左心室流出道梗阻也是该病常见特征。组织学表现为心肌细胞肥大,排列紊乱伴间质纤维灶的形成。流行病学研究表明HCM的发病率约为1/500[12],可在婴儿到老人的任何一个阶段发病,大部分患者预后良好,可达正常预期寿命。Maron[13]对744例HCM进行的跟踪随访,平均随访了8年,患者年病死率为14%,主要的死亡原因为猝死(51%)、进展性心力衰竭(36%)和心房颤动等导致的脑卒中(13%)。HCM是最常见的年轻人早发心因猝死原因,特别是年轻运动员。2014年欧洲心脏病学会发表了新版HCM的诊治指南,更新了猝死分层及植入型心律转复除颤器(ICD)适应证,采用绝对危险因素模式,计算出HCM患者5年心源性猝死的具体数值以指导临床工作[14]。HCM病因复杂,分为遗传性和非遗传性两大类,遗传性又可分为家族性(75%)、遗传代谢缺陷性疾病(9%)、功能异常综合征(9%)及神经肌肉疾病(8%)等。
1990年发现了第一个致病基因MYH-7,编码β-肌球蛋白重链,随后约26个基因,1 400个突变位点被发现,大约有50%~70%的患者可以检测到基因突变[12]。2011年,美国节律学会/欧洲心脏节律学会颁布了《遗传性心脏离子通道病与心肌病基因检测专家共识》[15],建议应对HCM患者检测MYH7、MYBPC3、TNNI3、TNNT2、TPM1等基因,有研究发现80%以上的HCM是由于以上基因的相关位点突变所致。目前认为HCM大多是由编码心肌肌小节收缩体相关蛋白的基因突变所致,致病基因主要包括编码粗肌丝蛋白(MYH7,MYH6,MYL2,MYL3)、细肌丝蛋白(TNNT2,TNNI3,TPM1,ACTC,TNNC1)、装配蛋白(TTN,MYBPC3)的基因。新报道的致病基因Z蛋白相关基因(LBD3,CSRP3,TCAP,VCL,ACTN2,MYOZ2,ANKRD1)和钙离子相关蛋白基因(JPH2,CASQ2,PLN,CALR3RYR2,DTNA),在人群中突变频率低,主要发生于散发人群,与HCM发病关系不明确。也有报道示编码桥粒蛋白(DSC2,DSG2,DSP,PKP2,JUP),转化生长因子(TGFb-3),跨膜蛋白(TMEM43),细胞骨架蛋白(DMD,LDB3),线粒体(TAZ),中间丝(LMNA,DES)等基因也可导致DCM[12]。在基因型阳性的患者中,3%~5%的患者同时携带两个或多个突变[16-17]。HCM基因型和表型的关系至今仍未完全阐明,也具有遗传异质性,可以存在不完全外显,外显率随年龄增长而增加。既往有研究表明,MYH7突变发病年龄较早,临床症状较重,生命周期短,甚至发生猝死等恶性心血管事件[18],而MYBPC3突变患者主要表现为发病年龄晚、肥厚程度轻、猝死危险因素少以及预后较好等,但梗阻与否和治疗手段上无明显差异[19]。TNNT2基因的突变导致的心肌肥厚非常容易引发心源性猝死[19]。长期的随访研究发现MYBPC3、MYH7和TNNT2的突变携带者的预后比较,差异无统计学意义(P>0.05)。TNNI3突变患者主要表现为心尖部肥厚,基于这种形态学改变,心尖部HCM患者,TNNI3的基因突变有可能为主要的遗传学病因,而且某些突变在一些同时患有HCM和预激综合征(WPW)的患者中出现[20],但其结论尚需大样本流行病学及进一步研究证实。D175N是TPM1基因的热点突变,Garcfa-Castro等[21]对 120例HCM患者筛查时发现一家系3位携带同样突变D175N,表现为室间隔严重肥厚,临床表现呼吸困难、心绞痛等。
1.3 致心律失常型右心室心肌病
ARVC又称为致心律失常性右心室发育不全、右心室室壁瘤、右心室DCM、羊皮纸样心脏、右心室心肌病。它是一种遗传性心肌病,以右心室功能障碍和室性心律失常为特征。组织学特点是正常心肌组织逐渐被脂肪组织和纤维组织替代。临床表现有心律失常、心力衰竭、发作性晕厥或猝死。
家族性ARVC占50%以上[22],常呈常染色体显性遗传,也有常染色体隐性遗传的报道。迄今为止,以发现多个连锁位点,现认为ARVC是一种桥粒病(DSC2,DSG2,DSP,PKP2,JUP),桥粒功能异常是致病的最后通路,非桥粒基因(TMEM43,TGF-3,LAMRJ,)可能通过影响桥粒发挥作用[23]。而非家族性ARVC可能与心肌炎相关。
1.4 限制型心肌病
RCM是以心室充盈受限、单侧或双侧心室舒张容量减少、收缩功能和室壁厚度正常或接近正常、伴增生性间质纤维化为特征的心肌病,临床表现上与缩窄性心包炎难鉴别。据文献报道,RCM的发病与地域、种族、性别等因素相关,多发生于热带和温带地区,但未见大规模统计数据。我国上海、山东、云南、广西等均有散发病例报道。组织学上分为浸润性和非浸润性两大类,浸润性病变为全身疾病造成的心肌局部组织学改变,见于淀粉样变性的心肌间质淀粉样物质堆积,类肉瘤的心肌内肉瘤样物质浸润,血色病的心肌内含铁血黄素的沉积,糖原贮积病的心肌内糖原颗粒过度积聚等,非浸润性病变包括心肌内膜纤维化与嗜酸细胞性心内膜炎两种。在排除炎症浸润、糖原代谢病、弹力纤维增生症、淀粉样变性等继发因素后,考虑为特发性RCM,特发性RCM多见于儿童,与DCM和HCM相比,特发性RCM发病率最低,但进展快、预后最差,确诊后平均生存周期仅为2年。
目前认为RCM发病机制仍不清楚,有报道在特发性的RCM患者中,30%有家族史,遗传方式以常染色体显性遗传为主,也可表现为常染色体隐性遗传。现已发现结蛋白(DES)、肌节蛋白(TNNT2)、α-心肌肌动蛋白(ACTC)、抗凋亡蛋白BAG-3(BAG3)、α-半乳糖苷酶(GLA)、肌靶蛋白(MYPN)、转甲状腺素蛋白(TTR)、肌钙蛋白I(TNNI3)和β-肌球蛋白重链(MYH7)等基因突变均可引起RCM[24],后两种心肌蛋白的基因突变还会同时引起心肌肥厚和限制性的生理改变。
2 左心室心肌致密化不全
左心室心肌致密化不全又称为海绵状心肌、窦状心肌持续状态或胚胎样心肌等,是由于胚胎形成过程中心肌致密化过程停滞,导致心室腔内突出的肌小梁和左心室腔交通且深陷的小梁间隙,病变多累积左心室,伴或不伴右心室受累。临床表现从无症状到心律失常、心力衰竭、体循环栓塞甚至心源性猝死。左心室心肌致密化不全有明显的家族遗传倾向,以常染色体显性遗传为主,有时也表现为X连锁遗传和线粒体遗传,12%~50%的左心室心肌致密化不全患者有家族史,且常合并心脏畸形及其他遗传性疾病,目前约发现15个基因参与左心室心肌致密化不全的发生,包括DTN1,LDB3,ACTC1,MYH7,TNNT2,MIB1,PRDM16,TPM1,MYBPC3,TAZ,LMNA,DTNA,FKBP-12,SCN5A,LBD3[25-26]。
3 遗传代谢病和功能异常综合征
遗传代谢性疾病的发生率在活产儿约为1/2 500[27],种类繁多,其中的5%合并心肌病。部分患者中,遗传代谢物质蓄积引起的心肌病变为早期的主要表现。Nugent等[28]报道,儿童心肌病中遗传代谢性疾病占8.9%。遗传代谢性疾病的发病多与机体蛋白质、脂类及碳水化合物代谢或能量合成的酶缺陷有关,可导致代谢产物或能量供应不足、底物或中间毒性代谢物质贮积引起相关器常、线粒体病及溶酶体病等。目前已知引起心肌肥厚的主要遗传代谢性心肌病主要为(1)糖原累积病:Pompe病、糖原累积病Ⅲ型、Ⅳ型、Danon病、PRKAG2心脏综合征等;(2)脂肪酸代谢障碍:如原发性肉碱缺乏症,肉碱棕榈酰基转移酶Ⅱ型缺乏症,多种酰基辅酶A脱氢酶缺乏症等;(3)线粒体病:丙酮酸脱氢酶缺乏症,呼吸链复合物Ⅰ、Ⅱ、Ⅲ、Ⅳ和Ⅴ缺陷,肌阵挛性癫痢-破碎红色肌纤维综合征(MERFF),Barth综合征,Senger综合征等;(4)溶酶体病:黏多糖贮积病(MPS)Ⅰ型、黏多糖贮积病Ⅱ型、单唾液酸四己糖神经节苷脂(GM1)神经节苷脂贮积症、GM2神经节苷脂贮积症、代谢病、Fabry病等。与心肌病相关的遗传代谢病和功能异常综合征详见表1。
小儿遗传代谢型心肌病又以原发性肉碱缺乏症及糖原累积病最为常见。据文献报道,合并DCM的先天性代谢缺陷(inborn error of metabolism,IEM)型心肌病的以脂肪酸氧化障碍及线粒体病常见,合并HCM的IEM以Pompe病多见,合并心肌致密化不全型心肌病的IEM以Barth综合征多见,合并RCM的IEM以代谢病及Fabry病常见。但一类代谢缺陷与一类心肌病并非一一对应的关系,同一种代谢缺陷在疾病不同时期可出现不同的改变,不同的代谢缺陷也可到导致相似的心肌改变。Colan等[29]在总结了855例年龄<18岁的HCM患者临床资料显示,因为IEM的占74例(8.7%),其中以pompe病最多。Towbin等[30]报道例小儿DCM,在明确病因的患者中IEM占11%,其中线粒体病常见,其次为Barth综合征和肉碱缺乏症。傅立军等[31]在2012年报道了对75例DCM儿童采用串联质谱技术进行遗传代谢筛查,筛查出6例肉碱缺乏患者,在予以补充肉碱治疗后取得满意疗效。
脂肪酸氧化障碍是一组少见的常染色体隐性遗传疾病。因涉及不同的酶或转运蛋白,临床表现轻重不一,轻者平时无症状,重者可致死。心脏表现是脂肪酸氧化障碍常见的临床表现,仅次于肝脏表现,主要为心肌病、心律失常、心源性休克及猝死。心肌病见于约1/3的脂肪酸氧化障碍患者,可呈心室肥厚伴收缩功能减低。Bonnet等报道脂肪酸氧化障碍107例,主要呈心律失常的占22%。能够引起心肌病的脂肪酸氧化障碍中常见疾病为:原发性肉碱缺乏症、极长链酰基辅酶α脱氢酶缺乏症、长链羟酰基辅酶α脱氢酶缺乏症以及肉碱棕榈酰基转移酶Ⅱ缺乏症等。正常心肌能量主要由脂肪酸分解供应。长链脂肪酸必须在肉碱协助下从胞浆转运至线粒体内方能进行β氧化分解。原发性肉碱缺乏症是由于肉碱转运体缺陷导致尿中肉碱大量丢失,心肌、骨骼肌、脑等细胞内肉碱贮存减少,影响脂肪酸代谢。多在2岁以前出现症状,临床表现为低酮性低糖血症、Rye综合征、昏迷。既往认为心肌病变以DCM和HCM为主。文献报道,24例原发性肉碱缺乏症患者中,DCM占58%,HCM占29%,心内膜弹力纤维增生症占13%,心肌病变无明显特异性。极长链酰基辅酶α脱氢酶缺乏症是由于线粒体脂肪酸氧化第一步的关键酶缺陷所致,新生儿和婴儿早期发病,常有心肌受累,患儿病死率高,表现为低血糖、DCM和HCM、心包积液等。长链羟酰基辅酶α脱氢酶缺乏症的临床表现有低酮性低糖血症、乳酸酸中毒、骨骼肌肌病,也有呈HCM变者。肉碱棕榈酰基转移酶Ⅱ位于线粒体膜并参与长链脂肪酸的线粒体跨膜转运,肉碱棕榈酰基转移酶Ⅱ型缺乏症可伴心血管征象。Pompe病是溶酶体酸性α-葡萄糖苷酶先天性缺陷引起的常染色体隐性遗传病,又称糖原累积病Ⅱ型,心脏、肌肉和肝脏等多种器官可受累。婴儿期发病者多因酶活性完全缺乏,常见喂养困难,肌无力、运动发育迟缓、呼吸困难和巨舌等。15%~23%患儿的首发症状为心力衰竭及心肌肥厚并呈进行性加重,可导致左心室流出道梗阻,也有左、右心室壁均肥厚。该病可伴有血清肌酸激酶及乳酸脱氢酶浓度增高。近75%的婴儿型Pompe病于12个月内死产。
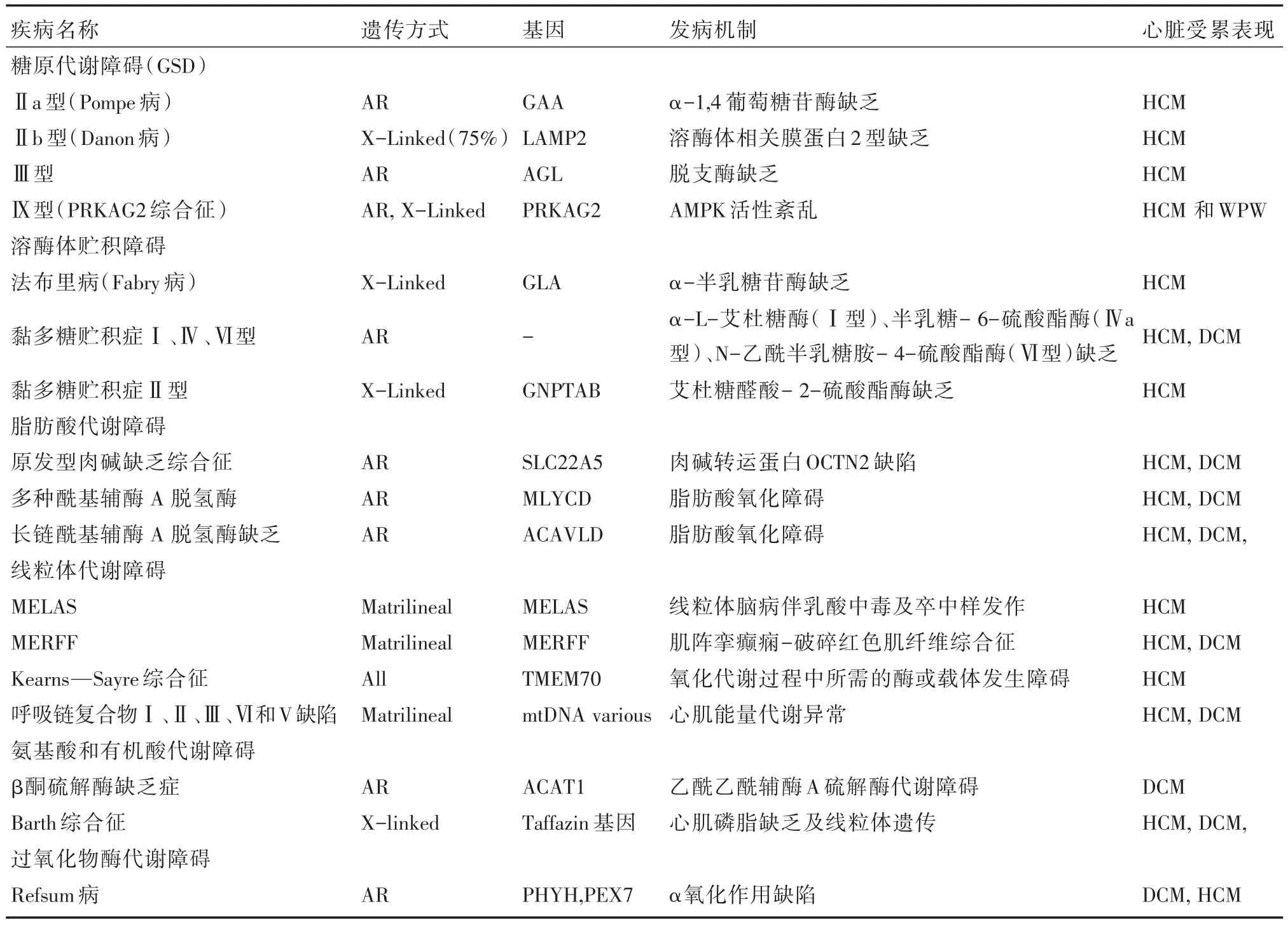
表1 与心肌病相关的遗传代谢病和功能异常综合征
与Pompe病相似,可引起心肌病的尚有Danon病。Danon病是X染色体上编码LAMP2基因突变所致的显性遗传病。由于溶酶体膜相关蛋白2缺乏导致糖原代谢障碍,累及心脏主要表现在心肌细胞内大量糖原储积引起,导致心室肥厚。该病常累及多系统病变,以心脏受累为主,临床上主要表现为心室肥厚。即可表现为肥厚性梗阻型心肌病,又可表现为于目前国内缺少Danon病的临床资料,且该病主要表现为心室肥厚而易误诊为HCM。近年有研究显示,与编码肌节蛋白基因突变引起的肥厚性心肌病相比,Danon病心脏受累可能有其自身特点[6]。Fabry病、线粒体心肌病等也各有不同的表现[32]。
4 神经肌肉疾病
与神经肌肉性疾病相关的心肌病患儿多数为编码dystrophy基因突变所致,为X-连锁遗传,包括Duchenne肌营养不良和Becker肌营养不良。典型的肌营养不良患者的肌活检可见肌纤维不同程度增大、变性坏死及结缔组织增生和脂肪浸润。肌营养不良不同程度累积心肌既可以表现为HCM也可以变现为DCM,患儿常在出现心功能能不全的临床表现之前数年,即可出现潜在的心电图异常或组织多普勒超声、心脏核磁发现心肌功能异常。目前发现10余种与Dystrophy基因变异热点[33-34]。
5 展望
遗传型心肌病的基因筛查目前已经在很多大型的商业实验室开展。基因检测不仅能更好地使人们了解该疾病的发病机理,还可以从分子水平为该病的诊断、筛查、预防和治疗提供参考。而建立致病突变和临床表型的联系,对于揭示心肌病的遗传特点、判断预后、指导治疗具有非常重要的意义。而越来越多基因型和表型关系的发现也为我们预防性的治疗提供了新的思路,如有猝死风险的基因携带者早期应用置入性心脏除颤器,可能会挽救他们的生命,而阻断突变基因引起的细胞信号的启动也应尽快由动物实验阶段转为临床研究。随着分子遗传学的发展,相信将有更多与心肌病有关的基因被发现,这将有利于我们对心肌病早期、准确的诊断,而将基因治疗用于临床,相信在有关技术不断完善的情况下也将实现。
[1]MARON B J,TOWBIN J A,THIENE G,et al.Contemporary definitions and classification of the cardiomyopathies:an American Heart Association Scientific Statement from the Council on Clinical Cardiology,Heart Failure and Transplantation Committee;Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups;and Council on Epidemiology and Prevention[J].Circulation,2006,113(14):1807-1816.
[2]ELLIOTT P,ANDERSSON B,ARBUSTINI E,et al.Classification of the cardiomyopathies[J].Kardiol Pol,2008,66(5):533-540,541-542.
[3]RUSSO A M,STAINBACK R F,BAILEY S R,et al.ACCF/HRS/AHA/ASE/HFSA/SCAI/SCCT/SCMR 2013 appropriate use criteria for implantable cardioverter-defibrillators and cardiac resynchronization therapy:a report of the American College of Cardiology Foundation appropriate use criteriatask force,Heart Rhythm Society,American Heart Association,American Society of Echocardiography,Heart Failure Society of America,Society for Cardiovascular Angiography and Interventions,Society of Cardiovascular Computed Tomography,and Society for Cardiovascular Magnetic Resonance[J].Heart Rhythm,2013,10(4):e11-e58.
[4]PINTO Y M,ELLIOTT P M,ARBUSTINI E,et al.Proposal for a revised definition of dilated cardiomyopathy,hypokinetic non-dilated cardiomyopathy,and its implications for clinical practice:a position statement of the ESC working group on myocardial and pericardial diseases[J].Eur Heart J,2016,37(23):1850-1858.
[5]HERSHBERGER R E,LINDENFELD J,MESTRONI L,et al.Genetic evaluation of cardiomyopathy-a Heart Failure Society of America practice guideline[J].J Card Fail,2009,15(2):83-97.
[6]DAUBENEY P E,NUGENT A W,CHONDROS P,et al.Clinical features and outcomes of childhood dilated cardiomyopathy:results from a national population-based study[J].Circulation,2006,114(24):2671-2678.
[7]BAIG M K,GOLDMAN J H,CAFORIO A L,et al.Familial dilated cardiomyopathy:cardiac abnormalities are common in asymptomatic relatives and may represent early disease[J].J Am Coll Cardiol,1998,31(1):195-201.
[8]KAMISAGO M,SCHMITT J P,MCNAMARA D,et al.Sarcomere protein gene mutations and inherited heart disease:a beta-cardiac myosin heavy chain mutation causing endocardial fibroelastosis and heart failure[J].Novartis Found Symp,2006,274:176-189,189-195,272-276.
[9]VILLARD E,DUBOSCQ-BIDOT L,CHARRON P,et al.Mutation screening in dilated cardiomyopathy:prominent role of the beta myosin heavy chain gene[J].Eur Heart J,2005,26(8):794-803.
[10] HERSHBERGER R E,HEDGES D J,MORALES A.Dilated cardiomyopathy:the complexity of a diverse genetic architecture[J].Nat Rev Cardiol,2013,10(9):531-547.
[11] HERMAN D S,LAM L,TAYLOR M R,et al.Truncations of titin causing dilated cardiomyopathy[J].N Engl J Med,2012,366(7):619-628.
[12] COATS C J,ELLIOTT P M.Genetic biomarkers in hypertrophic cardiomyopathy[J].Biomark Med,2013,7(4):505-516.
[13] MARON B J,OLIVOTTO I,SPIRITO P,et al.Epidemiology of hypertrophic cardiomyopathy-related death:revisited in a large non-referral-based patient population[J].Circulation,2000,102(8):858-864.
[14] ELLIOTT P M,ANASTASAKIS A,BORGER M A,et al.2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy:the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology(ESC)[J].Eur Heart J,2014,35(39):2733-2779.
[15] ACKERMAN M J,PRIORI S G,WILLEMS S,et al.HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies:this document was developed as a partnership between the Heart Rhythm Society(HRS)and the European Heart Rhythm Association(EHRA)[J].Europace,2011,13(8):1077-1109.
[16] VAN DRIEST S L,VASILE V C,OMMEN S R,et al.Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy[J].J Am Coll Cardiol,2004,44(9):1903-1910.
[17] VAN DRIEST S L,JAEGER M A,OMMEN S R,et al.Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy[J].J Am Coll Cardiol,2004,44(3):602-610.
[18] TAJSHARGHI H,FYHR I M.Structural effects of the slow/bcardiac myosin heavy chain R453C mutation in cardiac and skeletal muscle[J].Scand Cardiovasc J,2008,42(2):153-156.
[19]刘文玲,谢文丽,胡大一,等.十个汉族家族性肥厚型心肌病MYH7、MYBPC3和TNNT2基因筛查结果及相应的临床特征[J].中华心血管病杂志,2006,34(3):202-207.
[20] ELLIOTT K,WATKINS H,REDWOOD C S.Altered regulatory properties of human cardiac troponin I mutants that cause hypertrophic cardiomyopathy[J].J Biol Chem,2000,275(29):22069-22074.
[21] GARCIA-CASTRO M,COTO E,REGUERO J R,et al.Mutations in sarcomeric genes MYH7,MYBPC3,TNNT2,TNNI3,and TPM1 in patients with hypertrophic cardiomyopathy[J].Rev Esp Cardiol,2009,62(1):48-56.
[22] HAMID M S,NORMAN M,QURAISHI A,et al.Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria[J].J Am Coll Cardiol,2002,40(8):1445-1450.
[23] 仇晓亮,刘文玲,胡大一.致心律失常性右室发育不良/心肌病:从分子遗传学到临床[J].中国心脏起搏与心电生理杂志,2009,23(2):158-162.
[24] 黄红,计晓娟.临床诊疗限制型心肌病的研究进展[J].中国循环杂志,2015,30(6):594-596.
[25]刘欣,刘文玲.左心室心肌致密化不全心肌病研究进展[J].中国循环杂志,2016,31(2):198-200.
[26]柳胜华.左室心肌致密化不全型心肌病分子遗传机制研究[D].北京协和医学院,2013.
[27] APPLEGARTH D A,TOONE J R,LOWRY R B.Incidence of inborn errors of metabolism in British Columbia,1969-1996[J].Pediatrics,2000,105(1):e10.
[28]中华医学会儿科学分会心血管学组,编辑委员会中华儿科杂志.儿童心肌病遗传代谢性病因的诊断建议[J].中华儿科杂志,2013,51(5):385-388.
[29] COLAN S D,LIPSHULTZ S E,LOWE A M,et al.Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children:findings from the Pediatric Cardiomyopathy Registry[J].Circulation,2007,115(6):773-781.
[30] TOWBIN J A,LOWE A M,COLAN S D,et al.Incidence,causes,and outcomes of dilated cardiomyopathy in children[J].JAMA,2006,296(15):1867-1876.
[31]饶姣.儿童心肌病的遗传代谢病因分析及相关研究[D].南方医科大学,2014.
[32] WICKS E C,ELLIOTT P M.Genetics and metabolic cardiomyopathies[J].Herz,2012,37(6):598-611.
[33] DIEGOLI M,GRASSO M,FAVALLI V,et al.Diagnostic work-up and risk stratification in X-linked dilated cardiomyopathies caused by dystrophin defects[J].J Am Coll Cardiol,2011,58(9):925-934.
[34] COHEN N,MUNTONI F.Multiple pathogenetic mechanisms in X linked dilated cardiomyopathy[J].Heart,2004,90(8):835-841.
R542.2
A
1007-9688(2017)06-0815-06
10.3969/j.issn.1007-9688.2017.06.47
小儿肥厚型心肌病变的精准诊断及影像学新技术研究-2016年度广州市科技计划项目(项目编号:201607010055);胎儿及婴儿心血管疾病早期诊断及干预的策略和技术-2015年度广东省省级科技计划项目(项目编号:2015B070701008)。
张力力(1991-),女,硕士研究生,研究方向为儿童心血管病的诊断与治疗。
王树水,E-mail:wssxome@126.com
2016-12-01)
