1例2-甲基3-羟基丁酰辅酶A脱氢酶缺陷症患儿的临床诊治*
2017-12-28李莹莹崔亚利王霞戴维蒋冬梅张晓东王泓
李莹莹 崔亚利 王霞 戴维 蒋冬梅 张晓东 王泓△
(1.四川大学华西第二医院检验科;2. 妇儿疾病与出生缺陷教育部重点实验室,四川 成都 610041)
临床论著
1例2-甲基3-羟基丁酰辅酶A脱氢酶缺陷症患儿的临床诊治*
李莹莹1,2崔亚利1王霞1戴维1蒋冬梅1张晓东1王泓1△
(1.四川大学华西第二医院检验科;2. 妇儿疾病与出生缺陷教育部重点实验室,四川 成都 610041)
目的:通过对1例2-甲基3-羟基丁酰辅酶A脱氢酶缺陷症患儿的临床表现及检查结果的分析,提高对此类疾病的认识,为该病的及时诊断和治疗提供依据。方法采用回顾性方法对我院收治的这名患儿的临床症状和体征、实验室常规检查结果、血、尿串联质谱和气相质谱检查结果、X光胸片、彩超、头颅MR、脑电图以及诊疗情况进行回顾和分析。结果患儿3岁,主要临床表现初期以腹泻、呕吐为主,后来出现嗜睡、意识障碍,气促、难以纠正的代谢性酸中毒等。头颅MRI显示双侧侧脑室轻度扩张,脑沟、脑裂稍增宽。脑电图检查结果为界线性幼儿期脑电图,额、中央区尖波数次发放。血串联质谱检查天冬氨酸等氨基酸升高,尿气相质谱2-甲基-3-羟基丁酸明显增高,但未发现甲基巴豆酰甘氨酸和2-甲基乙酰已酸的异常,基因检测结果为hadh2基因第4外显子p.R130C突变。结论2-甲基3-羟基丁酰辅酶A脱氢酶缺陷症极为罕见,对有不明原因的难于纠正的代谢性酸中毒、高氨血症、或不明原因的精神萎靡、意识障碍等神经系统症状患儿应警惕该病,可送血或尿标本进行串联质谱、气相质谱及基因分析,以便早期诊断及治疗。
遗传代谢病;2-甲基3-羟基丁酰辅酶A脱氢酶缺陷症;串联质谱技术;气相质谱技术
2-甲基3-羟基丁酰辅酶A脱氢酶缺陷症(2-methy1-3-hydroxybutyryl-CoA ehydrogenase deficiency,MHBDD)是一种罕见的X连锁隐形遗传代谢性疾病,该病是由于异亮氨酸在分解过程中的2-甲基3-羟基丁酰辅酶A脱氢酶活性异常使2-甲基3-羟基丁酸在体液内蓄积,导致临床症状多样,且常在早期侵犯神经系统[1]。2000年Zschocke等[2]首次发现报告此病以来,全球报告例数不足30例[3],2013年,舒剑波等[4]对中国首例患者及家系基因突变进行了分析。因该病较少见,且其临床表现复杂,在临床诊断过程中容易误诊。
本文对我院收治的1例初期以腹泻、呕吐为主,后来出现嗜睡、意识障碍,气促、难以纠正的代谢性酸中毒等临床表现,最终确诊为2-甲基3-羟基丁酰辅酶A脱氢酶缺陷症患儿的临床诊治过程进行回顾性分析,为临床及时诊断治疗此种遗传代谢性疾病提供依据,避免误诊的发生。
1 病例资料
1.1 基本资料
患儿男,3岁,因吐泻3天,精神萎靡、气促1天,加重伴意识障碍1小时由外院转入我院儿科ICU,入院后多次查血气分析提示代谢性酸中毒,且难以纠正,行两次肾脏替代治疗(Continuous renal replacement therapy,CRRT)后,患儿代谢性酸中毒纠正,意识好转,气促缓解,第5天成功脱机。脱机后患儿意识模糊,吞咽功能差,不能竖颈,四肢肌力及肌张力低,于第8天开始行神经康复治疗。患儿为第一胎第一产,足月,出生时体重3.4 Kg,出生情况良好,母乳喂养,8个月开始添加辅食,生长、运动、语言、智力等发育正常,患儿父母否认不洁饮食及特殊药物服用史,否认毒物接触病史,否认传染病接触史。
体格检查:T36.7℃,P159次/分,R36次/分,BP102/62 mmHg。急性危重病容,自主体位,神志模糊,皮下无出血,全身无水肿,全身浅表淋巴结未扪及肿大。结膜正常,瞳孔等大等圆,左3 mm,右3 mm,对光反射正常。鼻翼有扇动,口唇红润,口腔黏膜正常,咽部充血,扁桃体I度肿大,未见疱疹及分泌物。颈阻阳性,呼吸运动对称,深大呼吸,三凹征阳性,呼吸音稍粗,未闻及干湿啰音。心律齐,心音有力,未闻及杂音。全腹柔软,肝脏肋下未触及,脾脏未触及, Babinski征阴性,Kernig征阴性。
1.2 临床诊断方法
该患儿入院诊断经血常规、血气分析、血糖、血氨、凝血功能筛查、降钙素原、血培养、痰培养、脑脊液检查、胸片、心脏彩超、串联质谱/气相质谱筛查等初步确诊,脑电图、头颅MRI了解颅内受累情况。
1.3 实验室检查
WBC 16.25×109·L-1↑,N 71.9%↑,L 24.3%,HB126 g·L-1,PLT 340×109·L-1,白细胞总数及中性粒细胞百分比增加提示有炎症感染;PH 7.040↓,PCO21.6 KPa↓,PO229.1 KPa↑,BE-25.4 mmol·L-1↑,HCO3-3.2 mmol·L-1↓,提示代谢性酸中毒,从患者多次检测结果看,酸中毒较难纠正;血氨40.3 μmol·L-1↑;PT 17.2 s↑,APTT 45.2 s↑,TT 17.6 s,DDI 0.64 mg·L-1,Fg 216 mg·dL-1;降钙素原2.55 ng·mL-1↑;血培养48 h及5 d均无细菌生长,排除败血症;痰培养、心肌损伤标志物、随机血糖、输血免疫、大便常规未见明显异常;我院两次手足口病原核酸检测未见异常,排除手足口病。脑脊液细胞学:有核计数3×106·L-1,余未见异常。脑脊液生化:脑脊液糖 4.6 mmol·L-1↑,脑脊液氯化物151.2 mmol·L-1↑,余未见异常。经过11天治疗后复查,血常规检查正常(WBC 6.6×109·L-1,N 42.8%、L 40.3%);肝肾功能及电解质无异常;降钙素原正常(0.02 ng·mL-1);血气分析酸中毒纠正(PH 7.499,PCO23.53 KPa,PO213.92 KPa,BE 2.525 mmol·L-1,HCO3-25.6 mmol·L-1)。
1.4 胸片
入院后第三天胸部正位X光显示双肺纹理增多模糊,提示双肺炎症,见图1。

图1 胸部正位X光片:显示双肺纹理增多模糊
1.5 彩超
心脏超声检查显示形态结构未见明显异常,左心收缩功能测值正常;肾脏超声检查双肾、输尿管、膀胱均未见异常。
1.6 血串联质谱检测
结果显示甘氨酸(Glycine,Gly)1263.82↑,亮氨酸(Leucine,Leu)381.49↑,鸟氨酸(Ornithine,Orn)424.65↑,苯丙氨酸(Phenylalanine,Phe)120.86↑,游离肉碱(Free carnitine,C0)61.17↑,丙酰基肉碱/乙酰基肉碱(C3/C2) 0.28↑,丙二酰肉碱/3-羟基丁酰肉碱(C3DC/C4OH) 0.55↑,异戊酰烯基肉碱:1(C5:1) 0.23↑,游离肉碱/( 棕榈酰肉碱+十八碳酰肉碱) C0/( C16+C18) 61.17↑,结合患者其它检测结果,提示患者可能B酮硫解酶缺乏症。
1.7 尿气相色谱质谱检测
第一次送检尿液3-羟基丁酸1440.13↑,乙酰乙酸1.22↑,2-甲基3-羟基丁酸4.37↑,3-羟基异戊酸34.68↑,提示酮症,β-酮硫解酶缺乏症待排;第二次送检尿液2-甲基-3-羟基丁酸6.85↑,但未发现甲基巴豆酰甘氨酸和2-甲基乙酰已酸的异常,提示治疗后的2-甲基-3-羟基丁酰辅酶A脱氢酶缺陷症可能性,可初步排除β-酮硫解酶缺乏症。
1.8 头颅磁共振成像
显示患儿双侧侧脑室轻度扩张,脑沟脑裂稍增宽,见图2。
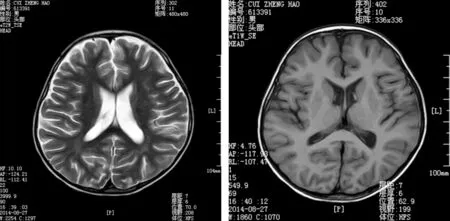
A B图2 头颅磁共振成像(MRI):显示患儿双侧侧脑室轻度扩张,脑沟、脑裂稍增宽注:A—T2加权像;B—T1加权像
1.9 脑电图
患儿脑电图检查结果为界线性幼儿期脑电图,额、中央区尖波数次发放。
1.10 基因检测
患儿外周静脉血基因检测结果为hadh2基因第4外显子p.R130C突变。
1.3 治疗方法
入院后立即予以呼吸机辅助通气、碳酸氢钠纠酸,美平抗感染,地塞米松抗炎、米力农强心、甘露醇及速尿利尿、新鲜冰冻血浆纠正凝血功能、丙球支持、维生素k1预防出血,磷酸肌酸钠保心,复合辅酶保护重要脏器、左卡尼汀及辅酶Q10、纠正电解质紊乱等对症支持治疗;遗传代谢性疾病筛查结果返回后,予以新朗欧抗感染、神经节苷酯及B族维生素营养神经、磷酸肌酸钠营养心肌等治疗,治疗后患者情况缓解后出院。
2 讨论
2-甲基3-羟基丁酰辅酶A脱氢酶缺陷症是近年来新发现的一种罕见的异亮氨酸代谢障碍性疾病,其临床表现主要累及神经系统,包括从一个静止性脑病到神经退行性改变[4]。Zschocke, Ensenauer[2-5]等报到2-甲基3-羟基丁酰辅酶A脱氢酶缺陷症患儿在新生儿及生长发育初期表现正常或存在轻微的发育落后,随后出现明显的精神发育迟滞。舒剑波等[4]报到病例曾于出生后第二天及2个月先后两次以肺炎住院治疗,1岁1个月该患儿因精神发育迟缓就诊。本例患儿3岁,出生情况良好,生长发育正常,起病初期为呕吐、腹泻、精神萎靡,外院诊断为脑炎、酸中毒进行治疗,但病情未好转气促进一步加重,转入我院后,血气检查结果分析提示代谢性酸中毒,且难以纠正,血氨明显增高,胸片显示双肺炎症,再进行两次连续性肾脏替代治疗(CRRT)后,患儿代谢性酸中毒纠正,意识好转,气促缓解,但患儿仍存在意识模糊,吞咽功能差,四肢肌力及肌张力低。从诊疗过程分析该类患者很容易被误诊为脑炎、肺炎等,而忽略为遗传代谢性疾病。另外,本病例在早期生长发育阶段均无异常表现,发病中有难以纠正的代谢性酸中毒,这与部分文献报到的临床表现有不同之处[2,4]。
2-甲基3-羟基丁酰辅酶A脱氢酶缺陷症的临床表现与β-酮硫解酶缺乏症(Beta-Ketothiolase deficiency,BKT)有相似之处,在临床诊断中不易区分[6]。BKT是一种少见的异亮氨酸代谢异常的常染色体隐性遗传性疾病,其缺陷酶是乙酰乙酰辅酶A 硫解酶,在异亮氨酸代谢中的异常产物与大量的肉碱结合从尿中排出,而过量的中间产物积聚, 可导致代谢异常及脏器功能异常,严重者可致精神运动发育迟滞。BKT以血或尿液中异戊酞肉碱( C5)及3-羟基异戊酞肉碱( C5-O H)异常增高为主要特征,尿气相色谱-质谱测定有2-甲基-3羟基丁酸、2-甲基乙酰乙酸、甲基巴豆酰甘氨酸增高,可以此作为BKT诊断依据。而典型的2-甲基3-羟基丁酰辅酶A脱氢酶缺陷症病人尿代谢谱为2-甲基-3羟基丁酸、甲基巴豆酰甘氨酸增高,但无2-甲基乙酰乙酸异常排出。本病例遗传代谢性疾病筛查送检有机酸明显升高,3-羟基异戊酰肉碱(C5-OH)、异戊烯酰肉碱(C5:1)显著增高,尿气相质谱第一次送检有3-羟基丁酸、乙酰乙酸、2-甲基3-羟基丁酸、3-羟基异戊酸增高,病人治疗后第二次送检2-甲基3-羟基丁酸明显增高,但无甲基巴豆酰甘氨酸及2-甲基乙酰乙酸的异常排出,故考虑为2-甲基3-羟基丁酰辅酶A脱氢酶缺陷症。
目前,国外有文献报道共发现有7种突变与2-甲基3-羟基丁酰辅酶A脱氢酶缺陷症相关,其基因为hadh2基因且均为错义突变,其中p.R130C是目前发现的最常见的突变[7-10]。舒剑波[4]等首次报道了2-甲基3-羟基丁酰辅酶A脱氢酶缺陷症患儿的家系基因突变分析,患儿是p.R130C突变,而该患儿的母亲为该位点的杂合子,证实该突变位点也存在于中国人群中。
2-甲基3-羟基丁酰辅酶A脱氢酶缺陷症的治疗采用限制异亮氨酸饮食疗法,并应积极的防治感染,从而减少线粒体应激反应,维护线粒体的稳态平衡,并给予维生素和辅因子治疗[4,11]。首先对患儿进行抗炎抗感染治疗,并以呼吸机辅助通气、碳酸氢钠纠酸及两次连续性肾脏替代治疗纠正电解质紊乱,另给予新朗欧抗感染、神经节苷酯及B族维生素营养神经、磷酸肌酸钠营养心肌等治疗。出院时情况明显好转,无发热、抽搐、意识障碍、咳嗽、吐泻等,精神食欲可,又经多日神经康复治疗后出院。
1 Zschocke J. HSD10 disease: clinical consequences of mutations in the HSD17B10 gene[J]. J Inherit Metab Dis, 2012, 35(1): 81-89.
2 Zsehocke J, Ruiter JP, Brand J, et a1. Progressive infantile neurodegeneration caused by 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency: a novel inborn error of branched-chain fatty acid and isoleucine metabolism[J]. Pediatr Res, 2000, 48(6): 852-855.
3 Fukao T, Akiba K, Goto M, et al. The first case in Asia of 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency (HSD10 disease) with atypical presentation[J]. J Hum Genet, 2014, 59(11): 609-614.
4 舒剑波,张玉琴,姜淑贞等. 2甲基3羟基丁酰辅酶A脱氢酶缺陷症一家系基因突变分析[J]. 中华儿科杂志,2013,51(10):783-788.
5 Ensenauer R, Niederhoff H, Ruiter JP, et a1. Clinical variability in 3-hydmxy-2-methylbutyryl-CoA dehydrogenase deficiency[J]. Ann Neurol, 2002, 51(5): 656-659.
6 Korman SH. Inborn errors of isoleucine degradation: a review[J]. Mol Genet Metab, 2006, 89(4): 289-299.
7 Perez-Cerda C1, García-Villoria J, Ofman R, et a1. 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase (MHBD) deficiency: an X-linked inborn error of isoleucine metabolism that may mimic a mitochondrial disease[J]. Pediatr Res, 2005, 58(3): 488-491.
8 Garcia-Villoria J,Navarro-Sastre A,Fons C,et a1.Study of patients and carriei′s with 2-methyl-3-Hydroxybutyryl-CoA dehydmgenase(MHBD)deficiency: difficuhiesin the diagnosis[J]. Clin Biochem, 2009, 42(1-2): 27-33.
9 Ofman R, Ruiter JP, Feenstra M, et al. 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency is caused by multations in the HADH2 gene[J]. Am J Hum Genet, 2003, 72(5): 1300-1307.
10 Rauschenberger K, Schöler K, Sass JO, et a1. A non-enzymatic function of 17beta-hydroxysteroid dehydrogenase type 10 is required for mitochondrial integrity and cell survival[J]. EMBO Mol Med, 2010, 2(2): 51-62.
11 Matern D, He M, Berry SA, et al. Prospective diagnosis of 2-methylbutyryl-CoA dehydrogenase deficiency in the Hmong population by newborn screening using tandem mass spectrometry[J]. Pediatrics, 2003, 112(1 Pt 1): 74-78.
Clinicaldiagnosisandtreatmentof2-methyl-3-hydroxybutyryl-CoAdehydrogenasedeficiencyinoneinfant:acasereport
Li Ying-ying1,2, CuiYa-li1, Wang Xia1, Dai Wei1, Jiang Dong-mei1, Zhang Xiao-dong1, Wang Hong1△
(1.Department of Laboratory Medicine, West China Second University Hospital, Sichuan University;2. Key Laboratory of Obstetric & Gynecologic and Pediatric Diseases and Birth Defects of Ministry of Education,West China Second University Hospital, Sichuan University, Sichuan Chengdu 610041)
Objective: To The clinical manifestations and examination results of one infant with 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency (MHBDD) was reviewed to enhance the understanding, diagnosis and treatment of MHBD.MethodsThe clinical manifestations and results of laboratory tests, tandem mass spectrometry of the blood sample, gas chromatography-mass spectrometry of the urine sample, chest X-ray, color Doppler examination, brain MRI and EEG as well as the treatment of this case were analyzed retrospectively.ResultsThis infant, aged 3 years old, mainly presented the symptoms of diarrhea and vomiting initially, and later lethargy, disturbance of consciousness, dyspnea and incorrigible metabolic acidosis. Brain MRI indicated mild ventriculomegaly and widening of sulci bilaterally. Borderline EEG was observed, with several sharp wave discharges in the frontal and central regions of the brain. Tandem mass spectrometry of the blood sample indicated increased levels of aspartic acid and other amino acids; gas chromatography of the urine sample indicated an obvious increase of 2-methyl-3-hydroxybutyric acid, but the levels of 3-methylcrotonyl-glycine or 2-methyl-acetoacetic acid were basically normally. Gene detection revealed p.R130C mutation in exon 4 of the hadh2 gene.ConclusionAs a rare disease, MHBDD may be suspected for infants presenting with incorrigible metabolic acidosis and hyperammonemia or neurological symptoms such as lethargy and disturbance of consciousness for unknown reasons. Examination techniques including tandem mass spectrometry, gas chromatography-mass spectrometry of the blood and urine samples and gene detections can be used for early diagnosis and treatment.
Inherited metabolic disease; 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency; Tandem mass spectrometry; Gas chromatography-mass spectrometry
四川大学横向课题(编号:15H0912)
李莹莹,女,技师,主要从事生化及遗传代谢病的临床检测,Email:272317706@qq.com。
△通讯作者:王泓,女,主管技师,主要从事遗传代谢病的临床检测与诊断,Email:1226203999@qq.com。
2017-8-26)
