36例Gilbert综合征临床特征分析
2017-12-28邓仲端程恒辉沈桂芬
李 咏,马 科,邓仲端,程恒辉,沈桂芬
华中科技大学同济医学院附属同济医院 1.内科; 2.感染科; 3.病理科,湖北 武汉 430030
36例Gilbert综合征临床特征分析
李 咏1,马 科2,邓仲端3,程恒辉3,沈桂芬1
华中科技大学同济医学院附属同济医院 1.内科; 2.感染科; 3.病理科,湖北 武汉 430030
目的通过对Gilbert综合征临床病例进行回顾性分析,以提高对该病的诊疗水平。方法回顾性分析华中科技大学同济医学院附属同济医院Gilbert综合征患者的临床资料,包括辅助检查、肝穿病理检查、基因检测结果。结果男性患者明显多于女性(86%vs14%),患者行饥饿试验后总胆红素(TBIL)、间接胆红素(IBIL)明显升高[TBIL:(39.77±17.88)μmol/Lvs(88.97±36.14)μmol/L,P<0.001;IBIL:(30.32±17.47)μmol/Lvs(80.61±33.76)μmol/L,P<0.001],行鲁米那诊断性治疗试验后TBIL、IBIL明显降低[TBIL:(107.81±28.24)μmol/Lvs(36.67±13.47)μmol/L,P<0.001;IBIL:(99.44±27.67)μmol/Lvs(28.20±13.89)μmol/L,P<0.001],基因测序结果为UGT1A1基因启动子碱基序列插入突变和UGT1A1基因第1号外显子突变,患者肝穿刺病理检查多正常或呈慢性炎症改变。结论Gilbert综合征以慢性间断性IBIL升高为主要特征,进行鲁米那试验、饥饿试验、病理检查可协助诊断,行基因测序可确诊,大部分患者无需特殊治疗,预后良好。
Gilbert综合征;总胆红素;间接胆红素;基因测序;肝穿病理检查
Gilbert综合征是一种遗传性疾病,临床表现为间断发生的以间接胆红素升高为主的非溶血性黄疸,一般无需特殊治疗,多见于青壮年,1901年由Gilbert和Leeboullet首先发现并报道。研究报道发病率高低不等,多在3%~10%[1-3]。在我国,Gilbert综合征也属于临床上黄疸查因中的一部分较难诊断的疾病,临床上表现为慢性、间歇性、非溶血性、IBIL升高,但大部分患者无明显症状,部分患者可有皮肤黏膜轻度黄染,或伴有恶心呕吐、乏力、纳差、肝区不适、脂肪不耐受等非特异性症状,一般预后良好,不需要药物治疗或长期监测。但临床上由于医师对其认识不足常被误诊为肝细胞性黄疸、梗阻性黄疸或溶血性黄疸,甚至肿瘤性疾病而进行广泛撒网式筛查,给患者带来不必要的负担,故在临床中对该病给予准确及时的诊断尤为重要。本文主要采用回顾性分析的方法,对2011年至2016年在华中科技大学同济医学院附属同济医院内科和感染科确诊为Gilbert综合征的36例患者的资料进行分析,旨在提高对该病的认识,以期鉴别临床胆红素升高相关疾病。
1 资料与方法
1.1一般资料通过饥饿试验、鲁米那治疗试验、临床肝穿刺病理诊断及基因测序诊断Gilbert综合征36例,其中合并乙肝患者7例,行病理活检者9例,行基因测序者9例,7例患者住院期间经鲁米那治疗并复查胆红素。在36例患者中,男31例,女5例,年龄(31.97±12.35)岁(14~64岁),TBIL(40.48±17.88)μmol/L(17.2~80.2 μmol/L),DBIL(9.67±5.14)μmol/L(4.2~30.5 μmol/L),IBIL(30.81±17.46)μmol/L(11.1~71.9 μmol/L)。谷丙转氨酶(25.71±19.01)IU/ml(7~98 IU/ml),谷草转氨酶(22.89±9.51)IU/ml(10~53 IU/ml)。
1.2诊断依据(1)依据病史、体征、实验室检查:黄疸以IBIL升高为主,DBIL可升高或正常;(2)临床症状较轻;(3)血常规、自身免疫性肝炎抗体、PNH检测、游离血红蛋白检测、葡萄糖-6-磷酸酶(G6PD)活性检测、红细胞渗透脆性实验、异丙醇实验等检查无异常,排除肝脏其他器质性病变;(4)结合饥饿/或利福平试验或诊断性鲁米那治疗试验;(5)肝组织病理学检查,组织学结构基本正常或呈慢性炎症改变;(6)基因诊断。
1.3纳入及排除标准纳入标准:符合上述Gilbert综合征主要诊断标准且住院资料完整。排除标准:不符合上述诊断标准,资料不完整、不确切,或可疑诊断,或合并其他疾病的病例。
1.4基因测序36例患者中,9例患者行基因测序检查,DNA提取和UGT1A1(尿苷二磷酸葡糖苷酸基转移酶)基因检测由华中科技大学附属同济医院心血管分子诊断中心完成。
1.5肝脏活检36例患者中,9例患者行肝脏活检,标本送我院病理科进行分析。
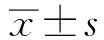
2 结果
2.1饥饿试验后,患者TBIL、IBIL的变化35例患者行饥饿试验中,行饥饿试验后,患者TBIL、IBIL明显上升,TBIL:(39.77±17.88)μmol/Lvs(88.97±36.14)μmol/L(P<0.001);IBIL:(30.32±17.47)μmol/Lvs(80.61±33.76)μmol/L(P<0.001)(见图1)。
2.2部分患者经鲁米那治疗后,TBIL、IBIL的变化7例患者经鲁米那治疗后,TBIL、IBIL明显下降,TBIL:(107.81±28.24)μmol/Lvs(36.67±13.47)μmol/L(P<0.001);IBIL:(99.44±27.67)μmol/Lvs(28.20±13.89)μmol/L(P<0.001)(见图2)。
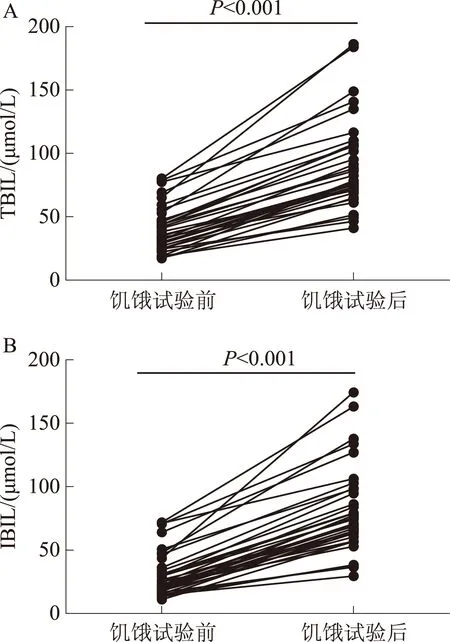
图1 饥饿试验前后TBIL、IBIL比较Fig 1 Comparison of TBIL and IBIL before and after the starvation test
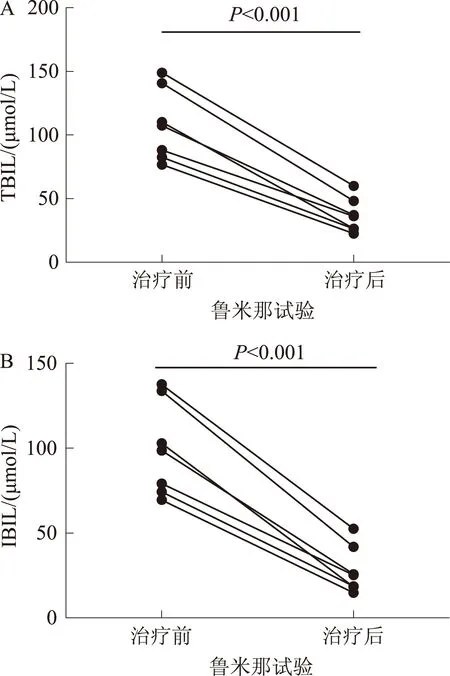
图2 鲁米那治疗前后TBIL、IBIL变化比较Fig 2 Comparison of the changes of TBIL and IBIL before and after the treatment by Rumina
2.3患者行基因检测后的结果9例患者行基因测序检查,5例为UGT1A1基因启动子TA盒中TA碱基序列插入突变(见图3A),3例为UGT1A1基因第一号外显子c.211G>A杂合突变(见图3B);1例为UGT1A1基因启动子TA盒中TA碱基序列插入突变合并p.Pro229Gln杂合突变(见图3C)。

图3患者基因测序(启动子、外显子区)结果A:UGT1A1启动子区(TA)n变异:患者为A(TA)7TAA/A(TA)7TAA基因型,正常人为A(TA)6TAA/A(TA)6TAA基因型;B:患者UGT1A1基因第一号外显子存在c.211G>A杂合突变,导致氨基酸p.Gly71Arg错义突变;C:患者同时存在UGT1A1基因TA碱基序列插入突变和第一号外显子氨基酸p.Pro229Gln错义突变
Fig3Resultsofpatientgenesequencing(promoter,exon) A: UGT1A1 promoter region (TA) n mutation; B: the first exon of UGT1A1 gene in patients with c.211G>A heterozygous mutation, resulting in missense mutations of amino acid p.Gly71Arg; C: UGT1A1 gene TA sequence insertion and the first exon amino acid p.Pro229Gln missense mutation
2.4部分患者肝穿刺病理检查结果肝小叶内中央静脉周围肝细胞脂肪变性,部分区域肝窦内淋巴细胞聚集,汇管区慢性炎性细胞浸润(见图4)。

图4 部分患者的肝穿刺病理检查结果 A:肝组织病理切片(200×);B:肝组织病理切片(400×)Fig 4 Pathological findings of liver biopsy in some patients A: liver pathological section (200×); B: liver pathological section (400×)
3 讨论
Gilbert综合征是由于遗传或后天获得性葡萄糖醛酸转移酶活性缺失或降低,导致肝细胞摄取IBIL障碍所致的疾病。根据现有文献[4-5]报道,发病年龄以中青年多见,男性发病率较高,比例为10∶1。本研究中,确诊为Gilbert综合征的患者也是以男性患者居多。临床上,诊断Gilbert综合征之前还需要完善相关检查以排除一些其他疾病,例如:PNH检测、游离血红蛋白检测、血红蛋白电泳、自身免疫性肝炎抗体、G6PD活性检测、红细胞渗透脆性实验、异丙醇实验等检查。怀疑为Gilbert综合征时,可进一步行饥饿试验,饥饿试验对该病的敏感性约80%,特异性极高。其原理是肝内胆红素配体和Z蛋白含量降低;血红素分解代谢升高;脂肪分解,导致游离脂肪酸增加,引起胆红素游离并释放入外周;同时肠蠕动减弱导致胆红素肠肝循环增加。每日400卡饮食的情况下,血浆IBIL增加>100%,或增加25.65 μmol/L,有诊断意义。本研究中,患者行饥饿试验后TBIL、IBIL升高明显,大多为2倍以上,饥饿试验阳性可以作为诊断Gilbert综合征的一个重要依据。除了饥饿试验,鲁米那治疗试验也是诊断该病的一个重要手段,鲁米那能够上调肝脏葡萄糖醛酸转移酶的活性,促进非结合胆红素与葡萄糖醛酸结合,降低血浆非结合胆红素的浓度[6]。本研究中,部分患者给予鲁米那试验性治疗并复查,多数患者胆红素明显下降,甚至可达正常,这也是我们诊断Gilbert综合征的一个重要依据。
对于目前Gilbert综合征的诊断而言,最重要的无疑是近年来飞速发展的基因测序诊断。Gilbert综合征是以UGT(尿苷二磷酸葡糖苷酸基转移酶)基因启动子区的基因多态性为遗传学基础[7],由于UGT酶蛋白的表达下调,肝脏内IBIL向DBIL的转化过程出现障碍,进而出现IBIL升高[8]。人体中仅有1种UGT即UGT1A1与胆红素代谢有关,目前,Gilbert综合征的基因诊断中,UGT1A1基因异常包括三种类型:(1)UGT1A1基因启动子TA盒中TA碱基序列插入突变,为常染色体隐性遗传。正常野生型纯合子A(TA)6TAA(TA)6TAA,简写为6/6;而纯合子患者为A(TA)7TAA(TA)7TAA,简写为7/7;杂合子为A(TA)6TAA(TA)7TAA。(2)发生在UGT1A1基因外显子区域的单碱基突变,包括第1号外显子第211位的鸟嘌呤(G)突变为腺嘌呤(A)导致编码产物中第71位甘氨酸变为精氨酸(Gly71Arg),第686位的胞嘌呤(C)突变为腺嘌呤(A)导致编码产物中第229位脯氨酸变为谷氨酞胺(Pro229Gln)等。突变可为纯合性、杂合性、复合杂合性。(3)近年来,人们在UGT1A1基因上发现了一个新的远端加强序列即苯巴比妥反应增强元件,该序列可存在T-G突变,引起转录活性显著降低。根据目前的报道[9-10],西方白人Gilbert综合征UGT1A1基因启动子区的TATA盒式结构的突变是其遗传基础,而亚洲人主要是基因的编码区发生了Gly71Arg的基因突变,高加索人患者常见的是A(TA)8TAA型的杂合子基因突变。对于我们的研究而言,大部分患者行测序后是前两种突变。
在回顾性研究中,部分患者行肝穿刺病理检查,多无明显异常。同时本研究中有7例患者合并慢性乙型肝炎,这部分患者大多因为长期IBIL较高,常规护肝治疗效果不佳,这些患者中,部分也同时进行了肝穿病理检查,也多提示肝脏炎症较轻,病理炎症分级多为G1S0、G1S1,较难解释长期黄疸异常这一现象。因此进行了Gilbert综合征相关检查,进而诊断为慢性乙型肝炎同时合并Gilbert综合征,明确了黄疸异常的原因。
本文通过回顾性研究,对于Gilbert综合征的诊断可归纳为以下几点:(l)重复多次肝功能检查均有间接胆红素血症;(2)血常规检查结果均正常,同时排除其他溶血相关疾病;(3)影像学检查:如腹部超声、CT、MRI、胆管造影等;(4)饥饿试验、鲁米那试验结果阳性;(5)多克隆UGT抗体的肝组织免疫组织化学检查,测定肝内UGT的活性程度或进行与GS相关的UGT基因启动子区TATAA序列的遗传学多态性测定;(6)排除肝脏其他器质性疾病,必要时肝脏穿刺病理活检。黄疸的鉴别诊断在临床上较为常见,临床医师应深入了解其发病机制,通过详细掌握病史和体检资料,有选择有序地安排各项实验和特殊检查以明确诊断。Gilbert综合征黄疸可持续终生,但大多数患者不需要特殊治疗,生存率也无影响,必要时可使用鲁米那短期治疗,同时积极剔除诱因,例如疲劳、饥饿、饮酒和感染等,教育患者养成良好的生活习惯。总之,随着目前基因测序技术的发展和临床医师诊断水平的提高,Gilbert综合征在临床上的确诊率越来越高,临床中也需要不断加强对该病的认识以便与其他黄疸行鉴别诊断,以期提高临床诊治水平。
[1] 陈濒珠, 林果为. 实用内科学[M]. 北京: 人民卫生出版社, 2009: 1827-1828.
[2] 李娟, 曲金宁, 张云丽, 等. Gilbert综合征8例临床特征分析及文献复习[J]. 肝脏, 2015, 20(2): 129-131.
LI J, QU J N, ZHANG Y L, et al. Analysis of clinical characteristics of eight patients with Gilbert syndrome [J]. Chinese Hepatology, 2015, 20(2): 129-131.
[3] 刘凡, 车芳, 骆子义. 利用多重荧光定量PCR协助诊断Crigler-Najjar综合征Ⅱ型及Gilbert综合征[J]. 胃肠病学和肝病学杂志, 2016, 25(12): 1389-1392.
LIU F, CHE F, LUO Z Y. Using multiple fluorescence PCR technique to assist in the diagnosis of Crigler-Najjar syndrome type Ⅱ and Gilbert syndrome [J]. Chin J Gastroenterol Hepatol, 2016, 25(12): 1389-1392.
[4] 肖文斌, 刘玉兰. 先天性高胆红素血症的分子遗传学研究进展[J]. 实用医学杂志, 2002, 18(2): 205-206.
[5] FRETZAYAS A, MOUSTAKI M, LIAPI O, et al. Gilbert syndrome [J]. Eur J Pediatr, 2012, 171(1): 11-15.
[6] 毛莉萍. Gilbert综合征(附2例报告)[J]. 齐鲁医学杂志, 2008, 23(1): 76-77.
[7] SAMPIETRO M, IOLASCON A. Molecular pathology of Crigler-Najjar type I and Ⅱ and Gilbert's syndromes [J]. Haematologica, 1999, 84(2): 150-157.
[8] ARIAS I M, LONDON I M. Bilirubin glucuronide formation in vitro; demonstration of a defect in Gilbert's disease [J]. Science, 1957, 126(3273): 563-564.
[9] 宋华, 宋力. Gilbert综合征及其分子遗传学基础[J]. 国际儿科学杂志, 2010, 37(4): 422-424.
SONG H, SONG L. Gilvert’s syndrome and molecular genetics basis [J]. Int J Pediatr, 2010, 37(4): 422-424.
[10] 徐羽中, 陈群蓉, 孙顺昌, 等. 应用实时荧光定量PCR技术分析UGT1A1基因启动子区A(TA)nTAA多态性[J]. 国际检验医学杂志, 2016, 37(13): 1806-1808.
XU Y Z, CHEN Q R, SUN S C, et al. Analysis on A(TA)nTAA polymorphism of UGT1A1 gene promoter by fluorescence real-time quantitative PCR [J]. Int J Lab Med, 2016, 37(13): 1806-1808.
Analysisofclinicalcharacteristicsof36patientswithGilbertsyndrome
LI Yong1, MA Ke2, DENG Zhongduan3, CHENG Henghui3, SHEN Guifen1
1.Department of Internal Medicine; 2.Department of Infectious Diseases; 3.Department of Pathology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China
ObjectiveTo improve the diagnosis and treatment of Gilbert syndrome by retrospective analysis.MethodsClinical data of patients with Gilbert syndrome in the Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology were retrospectively analyzed, and the results of gene sequencing were analyzed.ResultsThe number of male patients was significantly larger than female patients (86%vs14%). Total bilirubin (TBIL) and indirect bilirubin (IBIL) of most patients were significantly increased after starvation test [TBIL: (39.77±17.88) μmol/Lvs(88.97+36.14) μmol/L,P<0.001; IBIL: (30.32±17.47) μmol/Lvs(80.61±33.76) μmol/L,P<0.001], TBIL and IBIL of most patients were significantly decreased [TBIL: (107.81±28.24) μmol/Lvs(36.67±13.47) μmol/L,P<0.001; IBIL: (99.44±27.67) μmol/Lvs(28.20±13.89) μmol/L,P<0.001]after phenobarbital treatment. UGT1A1 gene promoter sequence insertion mutation and UGT1A1 gene mutation in exon 1 were found in gene sequencing. Normal or chronic inflammation were observed in liver biopsy pathological examination.ConclusionGilbert syndrome mainly manifested as chronic and intermittent IBIL increase. Luminal test, starvation test and pathological examination are helpful in diagnosis, gene sequencing can diagnose the disease. Most patients have good prognosis without special treatment.
Gilbert syndrome; Total bilirubin; Indirect bilirubin; Gene sequencing; Liver biopsy
李咏,博士,主治医师,研究方向:感染性疾病。E-mail:51940774@qq.com
沈桂芬,博士,副主任医师,研究方向:感染性和自身免疫性疾病。E-mail:guifenshen@126.com
10.3969/j.issn.1006-5709.2017.12.021
R57
A
1006-5709(2017)12-1409-04
2017-01-08
李健)
