金属有机骨架材料固定生物大分子的研究进展
2017-12-22谷娜,李恒,赵远
谷 娜,李 恒,赵 远
(河北科技大学理学院,河北 石家庄 050018)
金属有机骨架材料固定生物大分子的研究进展
谷 娜,李 恒,赵 远
(河北科技大学理学院,河北 石家庄 050018)
金属有机骨架(MOFs)具有超高的比表面积、可调的孔径、多样的结构组成、开放的金属位点和化学可修饰等性能。近年来,MOFs材料作为稳定的、高效的、可重复使用的和廉价的生物大分子固定化载体越来越引起人们的研究兴趣。生物大分子-MOFs体系在改进生物催化剂的效率及可回收性、分子传感、药物输送和基因治疗等方面具有广阔的应用前景。讨论了生物大分子在MOFs载体材料上固定的方法和方式,生物大分子可以通过物理吸附或共价键作用固定在MOFs表面,或通过与配位基团发生亲水或疏水作用扩散进入MOFs孔道,或通过共价键或配位键包埋在其晶体结构中,介绍了相关研究进展及应用。设计具有大孔径的高介孔MOFs材料、设计不同的功能化MOFs材料及以环境友好的方式合成所需的生物大分子-MOFs体系等,可进一步扩大MOFs材料在生物大分子固定领域的应用范围。
金属有机骨架(MOFs);蛋白质;酶;核酸;生物大分子;固定化
1 前 言
金属有机骨架 (Metal-Organic Framework, MOFs) 材料又称作多孔配体聚合物(Porous Coordination Polymers, PCPs),是一类由无机金属离子或金属簇与可调控的聚合有机配体共同构筑的多孔杂化无机-有机晶体材料[1-2]。1995年美国化学家Yaghi等[3]报道了使用过渡金属钴离子(Co2+)和有机配体均苯三甲酸(H3BTC)反应获得了具有二维结构的CoC6H3(COOH1/3)3(NC5H5)2.2/3NC5H5MOF化合物。在MOFs分子中通过配位键可以组装成多功能的三维拓扑结构, 并且可以根据特定的需要调整MOFs材料的形态和功能[4]。MOFs材料具有孔隙率高、孔径可调、比表面积大、金属含量可调、晶体结构可调和有机配体多样的特点[5],适用于各种应用情况。在MOFs发展早期,基于其规则的多孔结构,MOFs主要应用于气体存储[6]和流体分离[7]。随着对MOFs材料的深入研究,MOFs的应用已经扩展到储能、传感器、磁性和电子器件、非均相催化和生物医学领域。MOFs大的比表面积、良好的孔隙率、多变的结构和丰富的可调节的官能团,使得客体分子很容易扩散到其有序的框架结构中,从而促进主体和客体化合物的相互作用[8],MOFs作为对客体敏感的生物分子的固定化基质越来越引起人们的研究兴趣[9]。不同的生物分子如DNA、酶和其他蛋白质已被固定在MOFs材料中并且得到了广泛的应用。本文主要对采用MOFs固定生物大分子的应用进展进行综述。
2金属有机骨架材料
MOFs是多孔的结晶有机-无机杂化材料,由金属离子和有机物配体两部分组成。许多种类金属离子已被开发利用,主要包括过渡金属、p区元素、碱土金属和锕系元素[10]。金属的配位数决定了可能形成的分子几何构型,如线性、T / Y形、平面方型、平面锥型、八面体、三角双锥、三角棱柱和五边形双锥[11],如图1所示。使用不同大小和结构的多齿有机配体,能够控制合成的框架的结构和性能[12]。最常用的有机配体包括羧酸、胺 、硝酸盐、磷酸盐和磺酸盐,其中,阴离子羧酸盐与金属离子有更高的结合性。

图1 重要代表性MOFs[11]Fig.1 Schematic representation of important reported MOFs[11]
大多数MOFs都是通过液相法合成的,混合金属盐和配体溶液或者将溶剂加入到固态金属盐和配体中。合成MOFs的方法有缓慢蒸发法/直接沉淀法、溶剂热法、微波辅助法、电化学法、机械化学法和超声波方法。缓慢蒸发法是合成MOFs传统的路线,大多数反应不需要任何外部能量[10],一般在室温即可进行,但这种方法的主要缺点是所需时间较长[13]。溶剂热法也是一种广泛使用的合成MOFs的方法。微波辅助法是一种快速合成MOFs的方法,大大降低了合成时间[14]。电化学法,不需要金属盐,利用有机配体和电极的混合物中阳极的溶解产生金属离子,可以连续合成MOFs晶体[15]。机械法在机械力作用下使用少量溶剂即可快速合成MOFs,调节溶剂的加入量可以获得不同结构的配位物[16]。超声波产生的空化作用会导致温度或压力条件突然上升或下降,形成热点促进MOFs 快速均匀晶化[17]。此外,还可以通过自组装(或逐层组装)的路线来合成MOFs 材料,更好地控制和构建MOFs的形貌[18]。
MOFs材料所拥有天然的结构、电学、光学、催化及磁性等性能,且易于功能化,分子中存在可调节的亲水或疏水基团,可与生物分子之间存在强烈的相互作用,使其成为有用的生物分子固定载体,在传感、工业催化和生物医学等方面得以应用。
3 MOFs固定蛋白质
固体载体和蛋白质之间可以通过不同类型的化学键连接,这种相互作用分为5类:物理吸附、共价键结合、交联、基体包埋和微胶囊化[19]。相互作用的形式取决于被固定化的蛋白质的性质、固体载体的性质及所需的应用领域[20]。酶是一种具有催化活性的蛋白质,MOFs固定蛋白质的研究大多集中在MOFs作为酶分子固定化载体的研究,其在生物催化领域有重要应用。
大多数MOF-酶体系主要是通过3种不同的方法来构建:表面固定、孔隙扩散和在MOF自身合成过程中进行原位包埋等。
3.1 表面固定化
具有较大比表面积的MOFs材料经表面修饰后很容易负载大量的酶。MOF表面功能化可以促进酶和MOF之间形成稳定的化学键。酶在MOF表面固定化可以采用两种方式进行[21]。一是通过表面修饰来活化或预先合成用于有机连接的官能团,酶可以在随后的化学反应过程中与功能化的MOF以共价形式连接(图2a)。另外,酶分子可以先与其它一些可以进入MOFs材料孔道内的小分子(例如染料)共轭结合,小分子染料进入MOFs的微孔,酶分子仍然在MOFs材料的表面(图2b)。MOF中的有机配体和小分子染料可以通过氢键或π-π键相互作用结合,有助于形成稳定的主-客体络合作用。
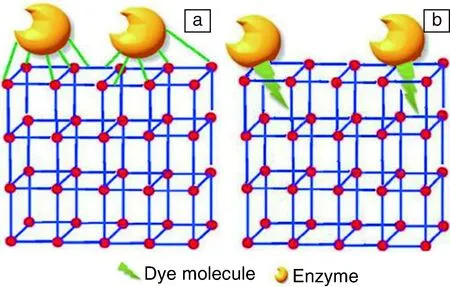
图2 酶表面固定化形式:(a)共价键结合;(b)染料标记的表面固定酶[21]Fig.2 Covalent attachment (a) and dye tagging (b) based surface anchoring of enzyme on the surface of MOF[21]
Jung和Park等[22]分别采用EDC活化In-基1D MOF、DCC活化2D([Zn(bpydc)(H2O)(H2O)]n;bpydc=2,2-bipyridine 5,5-dicarboxylate)MOF和3D(IRMOF-3)MOFs暴露在外面的羧基。活化的MOFs可与绿色荧光蛋白(EGFP)分子的氨基以共价键结合或通过酯化和酯交换反应将南极假丝酵母脂肪酶B(CAL-B)固定在MOF上。一维、二维和三维MOF负载EGFP能力分别为0.048,0.052和0.064 mg/g ,而负载CAL-B的能力分别为0.12,0.17和0.18 mg/g。与游离酶相比,固定化CAL-B酶活性增加了3倍并且可以循环利用。其中,CAL-B-IRMOF-3MOF复合物与游离酶相比显示出更好的催化活性,同时提高了IRMOF-3MOF的防水性,并且保持了其多孔性能。
Shih等[23]同样采用DCC交联剂活化法将胰蛋白酶(trypsin)固定在MIL-101(Cr)、MIL-88B(Cr)和MIL-88B-NH2上。在trypsin-MOF复合物中,胰蛋白酶中的氨基通过亲核反应与活化的羧基之间形成肽键。MOF中的胺基基团和DCC耦联剂的使用促进形成MOF-酶复合物。胰蛋白酶的分子大小大于MOF的孔径阻碍了胰蛋白酶向MOF孔道的扩散,因此保持了MOF的吸附能力。胰蛋白酶-MIL-88B-NH2复合物的催化活性在4个循环使用周期内比游离胰蛋白酶催化活性高69%。Trypsin-MIL-88B复合物可以通过离心法从生物成品回收,更利于产品的回收。然而,Shih等发现50%的乙腈水溶液洗涤液可能会使MOF-酶复合物中的羧基失活,导致酶从MOF表面流失而与载体分离。
Yu[24]及其合作者通过层层组装的方法在羧基功能化的Fe3O4纳米棒外包裹具有分级孔结构的3DMOF-100(Fe),制备了磁性Fe3O4@MIL-100(Fe)复合微球。 将Fe3O4@MIL-100(Fe)采用EDC/NHS或Zn2+活化后与Candida rugosa脂肪酶分子中的氨基以共价键或配位键相连,将酶固定在Fe3O4@MIL-100(Fe)上,固定酶后,明显改善了酶的循环使用性和稳定性,保持了酶的活性。
Qin和Liu等[25]在含有氨基的MOF(MIL-101(Al)-NH2)材料上,通过浸渍法吸附氯化高铁血红素蛋白,获得酶-MOF复合物。该酶-MOF复合物可以作为葡萄糖传感器使用,其上固定的氯化血红素蛋白质中的过氧化物酶可以用于比色法检测葡萄糖。
Ma等[26]报道了通过在ZIF-7、ZIF-8、ZIF-67、ZIF-68和ZIF-70上通过吸附法固定电催化剂甲基绿(MG)和葡萄糖脱氢酶(GDH)。由于疏水性作用、供-受体键和氢键的综合影响,ZIF-70表面吸附能力最强。在玻璃碳电极上通过滴落涂布MG-ZIF-70复合物,然后在MG-ZIF-70复合物上涂覆GDH制成葡萄糖生物传感器,其选择性检测葡萄糖的线性检测范围为0.1~2 mM。
Liu和Lin等[27]采用微波辅助方法先制备了携带异硫氰酸荧光素(FITC)的荧光素-胰蛋白酶共轭物,该共轭物中的FITC分子(分子大小0.9 nm×1.2 nm×1.4 nm)在涡旋离心作用力下,可以通过包埋方式进入铝基微孔MOF [Al(OH)(SDC)](CYCU-4)材料孔道内,从而获得MOF-酶复合体系,胰蛋白酶的负载量达到55.2 μg/mg。该生物催化体系具有良好的催化活性,蛋白水解效率达到47%,比游离的FITC-胰蛋白酶的消化效率43%高,并且水解时间由几个小时缩短至几分钟,该体系可重复使用至少5个周期。
Liu和Huang等[28]以小分子荧光染料4-氯-7-硝基苯并-2-氧杂-1,3-二唑(NBD)与胰蛋白酶结合得到胰蛋白酶-NBD,NBD与MOFs (MIL-101、DUT-4(Al)和UIO-66)之间有多个结合位点,因此对酶的负载能力增加。 其中,UIO-66负载胰蛋白酶-NBD的量达到80 μg/mg。
3.2 MOFs孔道的扩散
MOFs材料具有多孔结构,分子可以直接扩散到其孔道内,但是,由于大部分MOFs孔径小于2 nm,只有小于这个尺寸的化合物才可以扩散到其孔道内(图3)。

图3 蛋白质向 MOF孔道的扩散示意图[21]Fig.3 Schematic diagram of protein diffusion into the pores of MOF[21]
2006年,Pisklak等[29]将过氧化物酶(MP-11) 固定到多孔的纳米晶体Cu-MOF上。该Cu-MOF晶格中铜离子通过与4,4’-二苯基二羧酸连接形成铜二聚体,然后通过金属桥接配体形成平均孔径是1.78 nm的孔道。MP-11的大小为1.75 nm×3.3 nm,大于MOF孔道的大小,通过连续搅拌MOF-酶溶液,导致部分蛋白质链伸展,获得特定的取向,促进了MP-11向MOF孔道的扩散,负载量可以达到30 μmol/g。MP-11进入孔道后,在连续搅拌长达72 h的条件下,酶并没有表现出任何浸出现象。这可能是由于进入MOF框架内的酶不容易进行足够的展开,因此限制了其浸出。
Lykourinou等[30]在介孔TB-TATB MOF(孔径3.9 nm×4.7 nm)上固定MP-11。通过MOF颜色的变化及单晶体的吸收光谱证实了在室温条件下通过简单扩散的方式MP-11可以进入MOF孔道内,负载量达到19.1 μmol/g。与酶在二氧化硅上固定相比,这种MOF-酶载体具有更好的生物催化活性。由于酶进入MOF的孔道内,并且通过疏水作用与TB-TATB MOF相互作用,因此循环使用性较好,7次循环使用后,保留了约47%的催化活性。Lykourinou研究组[31]还研究了肌红蛋白(Mb)在Tb-TATB MOF孔道中的扩散,最大负载量可达到9.1 μmol/g。采用两种不同底物2,2-联氮-二(3-乙基苯并噻唑-6-磺酸)二铵盐(ABTS)和1,2,3 -三羟基苯(THB),研究了MOF的尺寸选择性。相对于MOF的孔径来说,ABTS是一种大分子,THB是小的分子,由于大分子底物不能到达Tb-meso MOF内部的Mb-活性位点,因此包埋在内部的酶对ABTS没有显示出过氧化氢酶的活性。与此相反,THB能够扩散到孔内,接近包埋的肌红蛋白酶,因此Mb-TB-TATB复合物对THB显示出催化活性。由于扩散缓慢,Mb@ TB-TATB催化活性仅为自由酶的一半,但这个生物催化体系可以循环使用15次。
Chen和Lykourinou等[32]还报道了亚铁血红蛋白Cyt c酶向纳米Tb-meso MOF孔道的扩散过程。如图4所示,尽管MOF的孔径小于Cyt c,蛋白质在MOF表面的物理吸附导致其部分展开从而发生构象的变化,使其最终迁移到MOF内部的孔隙。

图4 Cyt c酶在Tb-meso MOF孔道中的扩散机制示意图[32]Fig.4 Mechanism for the diffusion of Cyt c into the pores of Tb-meso MOF[32]
Rambabu等[33]将Cyt c酶扩散到基于2,6萘-二羧酸的Mn-MOF孔道内。该酶-MOF体系可用于检测导致水硬度的无机污染物硫酸根离子。
Deng 等[34]在合成MOF过程中使用多个含有苯环的有机配体,通过调节有机配体的数量,获得大孔径MOFs-74材料,最大孔径达到9.8 nm。 MOFs-74极高的孔隙率使得大分子蛋白质如Mb和绿色荧光蛋白(GFP)等可以在其中扩散。
Li和Farha[35]等将角质酶固定在具有分级孔结构的Zr基NU-1000,NU-1000具有直径为3.1 nm的六角形孔道和边缘长度为1.5 nm的三角型孔道,因此轴长为3.0 nm椭圆形的角质酶可以进入其孔道。固定化的酶可以用来水解p-硝基苯基丁酸盐 (PNPB),与自由酶活性相当,因为反应物和产物可以通过孔道扩散而与固定化的酶接触。
田运齐等[36]使用物理吸附的方法以大小为40~60 nm的HKUST-1纳米粒子组成的PD-meso MOF-3多级孔材料为载体对辣根过氧化物酶(HRP)进行了固定化。HPR分子尺寸大约为7.0 nm×4.4 nm×6.8 nm,可以进入HKUST-1纳米粒子构筑的PD-meso MOF-3的孔隙内,最大吸附量达到28.1 mg/g。在同样反应条件下,HPR固定化酶和原酶相比,催化过氧化氢和邻苯二胺生成2,3-二氨基酚嗪活性相当,且酶稳定性增强。
Huo等[37]先在油/水界面形成UIO-66/Fe3O4纳米复合物,而后采用琼脂凝胶法以磁性颗粒作为模板在UIO周围沉积ZIF-8层形成稳定的介孔微囊,可以封装CAL-B脂肪酶。
3.3 在MOF中酶的原位封装
大多数的MOFs孔隙直径小于酶的大小,因此需要另外一种MOF-酶体系扩大其应用范围。最近,研究者通过原位封装的方法在MOFs材料的框架内进行酶的包埋[31],如图5所示。采用这个方法的关键是在室温条件下合成MOF,以便在完成该过程后可以保持酶的活性和稳定性。

图5 酶在 MOF中进行原位封装[31]Fig.5 Schematic diagram of in-situ enzyme encapsulation within MOF[31]
Lyu等人[38]首先研究了这种方法。他们选择了一个化学和热稳定性好的MOF“ZIF-8”作为载体,并进一步提出了基于MOF-酶杂化体的自聚配位键。室温下在甲醇介质中慢速搅拌硝酸锌、2-甲基咪唑、Cytc(酶)和聚乙烯吡咯烷酮(PVP)混合物24 h获得 ZIF-8。PVP可以防止有机溶剂中的蛋白质聚集。获得的Cyt c@ZIF-8为菱形十二面体晶体,如图6所示。获得的Cyt c@ZIF-8和底物的结合力增加,由于酶上金属离子的激活作用,体系的催化活性比游离酶增加了10倍,这种生物催化载体被用于爆炸物的快速灵敏光学检测。使用上述方法还合成了HRP@ZIF-8、Cyt c@ZIF-10和 lipase@ZIF-8等酶-MOF复合物。

图6 Cyt c@ZIF-8的TEM 照片[38]Fig.6 TEM image of Cyt c@ZIF-8 crystal [38]
Shieh等人[39]室温下在水相中混合硝酸锌、咪唑、过氧化氢酶(CAT)和封端剂ICA 10 min ,实现在2 μm的ZIF-90单晶框架内固定CAT。固定化酶的催化活性低于天然形式,然而MOF的遮蔽效应可以提高酶的稳定性,从而有助于降低成本。
Liang等[40]研究了MOF晶体在酶上的生长机制。采用核磁共振光谱、原位同步小角X射线散射和ICP研究了在MOFs材料内不同蛋白质的仿生矿化过程。卵清蛋白(OV)、溶菌酶、HRP、血红蛋白、胰蛋白酶、脂肪酶、胰岛素、脲酶、核糖核酸酶、人血清白蛋白(HSA)和DNA都可以在MOFs材料(例如ZIF-8、HKUST-1、Eu-BDC、Tb-BDC和MIL-88A)的中心被矿化。这种生物矿化过程在保持酶的活性和稳定性的同时,有助于控制晶体生长。还可通过改变pH条件控制蛋白质的释放,在药物输送方面有潜在的应用。
Wu等[41]在25 °C水相溶液中直接混合葡萄糖氧化酶(GOx)、辣根过氧化物酶(HRP)、锌盐和2-甲基咪唑进行反应,在ZIF-8内同时进行GOx和HRP的原位包埋固定化,如图7所示。GOx/HRP/ZIF-8复合物可用于高效、灵敏、稳定地检测葡萄糖。酶包埋后MOF晶体结构完整,如图8 a和图8 b所示。HRP和GOx分别用荧光探针异硫氰酸罗丹明B(RhB,红光)和异硫氰酸荧光素(FITC,绿光)标记,图8 c的激光扫描共聚焦显微镜照片说明了酶在金属有机骨架内的位置。

图7 GOx/ HRP / ZIF-8复合物形成过程[41]Fig.7 Schematic diagram of GOx and HRP in-situ encapsulation within ZIF-8[41]

图8 ZIF-8晶体的TEM照片(a),GOx/HRP/ZIF-8复合物的TEM照片(b),复合物的激光扫描共聚焦显微镜照片(c) [41]Fig.8 TEM image of ZIF-8 crystal (a), TEM image of GOx/HRP/ZIF-8 composite (b), Confocal microscopic image of composite legend(c) [41]
Wu等[42]利用聚多巴胺(PDA)作为生物粘合剂,通过共价键或非共价键作用来束缚葡萄糖氧化酶-ZIF-8复合物,使其大小可以达到微米级。室温下,在PDA溶液中培养葡萄糖氧化酶/ZIF-8粉末24 h可以发生聚合反应,形成交联复合物。与游离形式葡萄糖氧化酶的相比,葡萄糖氧化酶/ZIF-8/PDA系统只表现出50%的催化活性。然而,固定形式的酶的活性可以保持更长的时间。上述体系在甲醇、乙醇和丙酮溶剂中,还表现出更好的稳定性,在10000 rpm的转速下离心可以回收利用复合材料。
Nadar等[43]以2-甲基咪唑、葡萄糖淀粉酶和醋酸锌为原料,采用自组装的一步法在室温合成了葡萄糖淀粉酶-ZIF-8复合物。包埋在ZIF-8内部的葡萄糖淀粉酶比自由酶的活性提高6倍,并且稳定性提高,6次循环使用时,活性还可达到57%,存储25 d活性仍为原来的91%。包埋在ZIF-8内部的酶二级结构发生改变,改进了催化活性。
表面固定法将酶通过物理吸附或共价连接附着在MOF表面,因其对酶的大小没有限制是最吸引人的方法。活性位点暴露在载体外可以增强酶的催化活性,降低蛋白质催化时间,从而减少酶的随机性,并使活性位点更容易到达底物[27]。使用简单的步骤将酶通过物理吸附固定在MOFs材料上,然而,如果酶与MOF载体的结合键相对较弱,这种酶很可能被洗掉。共价连接是解决这个问题的很好的选择,可以通过化学修饰的载体或通过小分子染料标记酶来增强结合键。
酶向MOFs孔道内扩散法中,酶包埋在MOFs材料孔道内,同时和MOF的配位基团发生亲水性或疏水性相互作用,有助于提高催化剂的稳定性[44],增加了材料的可重复使用性。但是因为大多数MOFs的孔道尺寸小于一些重要的酶,所以这种方法的使用范围有限,开发合成大孔径MOFs材料是应用这种方法的关键。
酶在骨架内的原位包埋可以通过共价键或配位键来实现。由于酶的完全包埋,这种固定化方法可以获得最大限度的稳定性,防止酶的浸出,可重复使用性较高,减少过程成本。另一个显著的优势是,这种方法对固定化酶的大小没有限制。那些尺寸大于MOF孔径的酶也可以通过这种方法来合成MOF-酶复合物。然而,由于底物向MOF的微孔内扩散的速度较慢或者底物分子大小大于MOF的孔径,使得底物不容易接触包埋在孔道内部的酶,因此,这种方法在某些情况下催化活性可能会比游离形式酶的低。能否采用这种方法取决于MOF和酶的结构参数,同时也需要考虑MOF-酶复合物的应用目标。在MOF中进行酶的原位封装时,要求MOF必需在室温条件下合成以便保持酶的结构和活性[38]。
4 MOFs固定核酸
使用MOF纳米载体还可以固定多聚核苷酸如DNA和RNA,可以保护其不被降解,便于其被细胞摄取。
Mirkin等[45]研究了基于新型UiO-66-N3(Zr6O4OH4(C8H3O4-N3)6)的核酸-MOF纳米配合物,采用二苯基环辛炔修饰的多聚核苷酸与叠氮化UIO-66反应,将DNA连接在纳米MOF表面,如图9所示。核酸-MOF纳米配合物空间和静电稳定性增加,对人类宫颈癌细胞渗透性增强,可用于基因治疗。

图9 MOF-DNA配合物形成过程[45]Fig.9 Schematic diagram of MOF-DNA conjugate [45]
He等[46]通过和金属位点配位的方式,将小干扰RNA负载在UiO型纳米MOF(锆-氨基三苯基二羧酸)表面,负载量30 μg/mL,可以保护SiRNA不被核酸酶降解,增加其被细胞摄取率。
5 结 语
MOFs材料可以作为稳定的、高效的、可重复使用且廉价的生物大分子的固定化载体。以MOFs为基础的生物大分子固定化系统已应用到生物催化、分子传感、治疗和诊断等领域。但是, MOF-生物大分子固定化体系的实际应用还处于初期阶段,近一步扩大MOFs材料的应用范围还有很多关键的问题需要解决,例如设计具有大孔径的高介孔MOFs材料、设计不同的功能化MOFs材料及以研发环境友好的方式合成所需的生物分子-MOFs体系等,不断提高MOFs-生物大分子固定化体系的商业化接受度。
References
[1] Rowsell J L C, Yaghi O M.MicroporousandMesoporousMaterials[J],2004,73(1-2):3-14.
[2] Zhou H C J, Kitagawa S.ChemicalSocietyReviews[J], 2014,43(16):5415-5418.
[3] Yaghi O M, Li G, Li H.Nature[J],1995,378(6558): 703-706.
[4] Cheetham A K, Rao C, Feller R K.ChemicalCommunications(Camb.) [J], 2006 (46):4780-4795.
[5] Férey G, Mellot-Draznieks C, Serre C,etal.Science[J], 2005,309(5743): 2040-2042.
[6] Loiseau T, Lecroq L, Volkringer C,etal.JournaloftheAmericanChemicalSociety[J], 2006,128 (31): 10223-10230.
[7] Britt D, Tranchemontagne D, Yaghi O M.ProceedingoftheNationalAcademyofSciencesoftheUnitedStatesofAmerica[J],2008,105 (33): 11623-11627.
[8] Farha O K, Hupp J T.AccountsofChemicalResearch[J],2010,43(8):1166-1175.
[9] Meek S T, Greathouse J A, Allendorf M D.AdvancedMaterial[J], 2011,23 (2): 249-267.
[10] Dey C, Kundu T, Biswal B P,etal.ActaCrystallographicaSectionB[J], 2014,70 (1): 3-10.
[11] Burnett B J, Barron P M, Choe W.CrystEngComm[J], 2012,14(11): 3839-3846.
[12] Kumar P, Bansal V, Deep A,etal.JournalofPorousMaterials[J], 2015, 22 (2): 413-424.
[13] Horcajada P, Chalati T, Serre C,etal.NatureMaterials[J], 2010,9(2): 172-178.
[14] Bux H, Liang F, Li Y,etal.JournaloftheAmericanChemicalSociety[J], 2009, 131(44):16000-16001.
[15] Martinez Joaristi A, Juan-Alcaňiz J, Serra-Crespo P,etal.CrystalGrowth&Design[J],2012, 12(7): 3489-3498.
[17] Jung D W, Yang D A, Kim J,etal.DaltonTransactions[J], 2010, 39(11):2883-2887.
[18] Lee D J, Li Q, Kim H,etal.MicroporousandMesoporousMaterials[J], 2012,163: 169-177.
[19] Wu X, Hou M, Ge J.CatalysisScience&Technology[J]2015,5 (12): 5077-5085.
[20] Cao L, Langen L V, Sheldon R A.CurrentOpinioninBiotechnology[J]. 2003, 14 (4): 387-394.
[21] Mehta J, Bhardwaj N, Bhardwaj S K,etal.CoordinationChemistryReviews[J], 2016,322 :30-40.
[22] Jung S, Kim Y, Kim S J,etal.ChemicalCommunications(Camb.) [J],2011,47 (10): 2904-2906.
[23] Shih Y H, Lo S H, Yang N S,etal.Chempluschem[J], 2012,77 (11): 982-986.
[24] Wang J Z, Zhao G H, Yu F Q.JournaloftheTaiwanInstituteofChemicalEngineers[J],2016,69 :139-145.
[25] Qin F X, Jia S Y, Wang F F,etal.CatalysisScience&Technology[J],2013,3(10): 2761-2768.
[26] Ma W, Jiang Q, Yu P,etal.AnalyticalChemistry[J],2013, 85 (15): 7550-7557.
[27] Liu W L, Lo S H, Singco B,etal.JournalofMaterialsChemistryB[J], 2013, 1(7): 928-932.
[28] Liu W L, Wu C Y, Chen C Y,etal.Chemistry-AEuropeanJournal[J], 2014,20(29):8923-8928.
[29] Pisklak T J, Macias M, Coutinho D H,etal.TopicsinCatalysis[J],2006,38(4): 269-278.
[30] Lykourinou V, Chen Y, Wang X S,etal.JournaloftheAmericanChemicalSociety[J],2011,133 (27): 10382-10385.
[31] Chen Y, Lykourinou V, Hoang T,etal.InorganicChemistry[J], 2012,51(17):9156-9158.
[32] Chen Y, Lykourinou V, Vetromile C,etal.JournaloftheAmericanChemicalSociety[J],2012,134 (32): 13188-13191.
[33] Rambabu D, Pradeep C P, Dhir A.JournalofMaterialChemistryA[J], 2014,2:8628-8631.
[34] Deng H, Grunder S, Cordova K E,etal.Science[J], 2012,336 (6084): 1018-1023.
[35] Li P, Modica J A, Howarth A J,etal.Chem[J], 2016, 1(1):154-169.
[36] Tian Yunqi (田运齐), Wu Xiaofang(吴小芳), Hou Wenying(侯文颖),etal.JournalofLiaoningNormalUniversity(NaturalScienceEdition)辽宁师范大学学报(自然科学版)[J],2016,39(3):373-376.
[37] Huo J, Marcello M, Garai A,etal.AdvancedMaterial[J], 2013,25(19):2717-2722.
[38] Lyu F, Zhang Y, Zare R N,etal.NanoLetters[J], 2014,14 (10): 5761-5765.
[39] Shieh F K, Wang S C, Yen C I,etal.JournaloftheAmericanChemicalSociety[J], 2015,137(13):4276-4279.
[40] Liang K, Ricco R, Doherty C M,etal.NatureCommunication[J],2015,6:7240.
[41] Wu X, Ge J, Yang C,etal.ChemicalCommunications(Camb.) [J], 2015,51(69):13408-13411.
[42] Wu X, Yang C, Ge J,etal.Nanoscale[J], 2015,7(45):18883-18886.
[43] Nadar S S, Rathod V K.InternationalJournalofBiologicalMacromolecules[J],2017,95:511-519.
[44] Pisklak T J, Macias M, Coutinho D H,etal.TopicsinCatalysis[J],2006,38 (4):269-278.
[45] Morris W, Briley W E, Auyeung E,etal.JournaloftheAmericanChemicalSociety[J], 2014,136 (20): 7261-7264.
[46] He C, Lu K, Liu D,etal.JournaloftheAmericanChemicalSociety[J], 2014,136 (14): 5181-5184.
Recent Advances in Biomacromolecules Immobilization by Metal Organic Frameworks
GU Na, LI Heng, ZHAO Yuan
(School of Science, Hebei University of Science and Technology, Shijiazhuang 050018, China)
Metal organic frameworks (MOFs) with properties of high specific surface area, tunable porosity, controllable structure, open metal sites and desirable functionality, have been demonstrated as stable, robust, reusable, efficient, and low-cost substrates for biomacromolecule immobilization, which have motivated increasing research interest. Biomacromolecule-MOFs composites have extensive application in improvement of biocatalyst efficiency and promising recyclability, molecular sensing, drug delivery and gene therapy. This review focuses on the progress in the application of MOFs as biomacromolecule immobilization matrix and the methods and modes for biomacromolecule immobilization to MOFs substrates. The biomacromolecule can be immobilized on the surface of MOFs by physical absorption or covalent bond, diffuse into the pore of MOFs by functional interactions with the MOF ligand moieties or be encapsulated within framework structure by covalent bond or coordination bond during mineralization of MOFs. The design of highly mesoporous and various functional MOFs and the eco-friendly synthesis processes of biomacromolecule-MOFs composites will enlarge the application range of MOFs as substrates for biomacromolecule immobilization.
metal organic frameworks (MOFs); protein; enzyme; nucleic acid; biomacromolecule; immobilization
2017-09-22
河北省自然科学基金资助项目(B2016208054)
谷 娜,女,1981年生,副教授, Email:
gunagao@qq.com
10.7502/j.issn.1674-3962.2017.11.04
TB34
A
1674-3962 (2017)11-0833-07
(编辑 惠 琼)
