Fe原子吸附对单层WS2结构和性质的影响
2017-12-21徐位云汪丽莉宓一鸣赵新新
徐位云 汪丽莉 宓一鸣 赵新新,*
Fe原子吸附对单层WS2结构和性质的影响
徐位云1,2汪丽莉1宓一鸣1赵新新1,*
(1上海工程技术大学基础教学学院,上海 201620;2上海工程技术大学材料工程学院,上海 201620)
本文采用基于密度泛函理论(DFT)的第一性原理方法研究了Fe原子吸附对单层WS2结构和性质的影响。研究结果表明:Fe原子吸附在W原子的顶位最稳定,相应的原子吸附能为1.84 eV。Fe与衬底间的相互作用削弱了紧邻W―S键,使其键长增大0.011 nm。由于衬底原子的影响,Fe原子轨道的电子重新分布,形成了2B左右的局域原子磁矩。在低覆盖度下(0.125和0.25 ML),磁性作用以超交换作用为主,铁磁序不稳定。而在高覆盖度下(0.5和1.0 ML),Fe原子间距减小,磁性作用以RKKY作用为主,铁磁序稳定。电子结构的计算结果显示,在高覆盖度下,Fe/WS2结构在费米能级处的电子自旋极化率等于100%。自旋向上与向下通道分别为间接带隙的半导体和金属。在1.0 ML覆盖度下,自旋向上的禁带宽度约为0.94 eV。这说明Fe原子吸附可以将直接带隙的WS2半导体转变成半金属,形成一种潜在的自旋电子器件材料。
密度泛函理论;Fe/WS2;过渡金属硫化物;交换作用;半金属
1 引言
近年来,具有类石墨烯结构的层状过渡金属硫化物(MX2;M = Mo、W等;X = S、Se、Te等)引起了人们的广泛关注1–4。体相过渡金属硫化物由X-M-X原子层堆积而成。层内由强M-X化学作用结合,层间由范德华力结合。过渡金属硫化物性质和原子层堆叠方式有很大关系。例如体相一般是间接带隙半导体,随着原子层厚度减小,单层时转变为直接能隙半导体5–7。二维结构和特殊的电子结构使过渡金属硫化物单层和多层薄膜在电子器件、光电器件和太阳能电池等领域有着非常诱人的应用前景5–7。
传统电子器件利用电子的电荷属性实现信息的存储和逻辑运算,利用电子的自旋属性设计和制造自旋电子器件是未来电子器件发展的潜在方案之一。半金属(half metal,HM)材料对于不同自旋的电子具有完全不同的电学性质,被认为是重要的自旋电子材料8–12。常规半金属材料往往是三维材料。一般由两种或两种以上元素组成,如三元金属化合物(NiMnSb、PtMnSb、FeVSb和NiTiSn)、过渡金属氧化物(CrO2和Fe3O4等)、钙钛矿型化合物(Sr2FeMoO6和Ca2FeMoO6)等13–19。
由于过渡金属硫化物本身没有磁性,利用一定的方法破坏时间反演对称性,引入磁性相互作用,有利于探讨过渡金属硫化物在自旋电子器件上的应用可能性。利用(3或4过渡金属)磁性原子掺杂和修饰是在非磁性材料中引入磁性相互作用是最直接的方法20–22。Ramasubramaniam等23采用密度泛函理论(DFT)和Monte Carlo方法研究了Mn掺杂的单层MoS2系统,研究结果表明该系统具有室温下的铁磁序,是潜在的稀磁半导体材料。Li等24研究了V、Nb和Ta掺杂WS2的电子结构和磁性,Nb和Ta掺杂的WS2体系表现出半金属性质。Zhao等25也证实过渡金属原子掺杂都可以在过渡金属硫化物单层中引入磁性。
本文采用密度泛函理论的第一性原理方法研究了过渡金属Fe原子吸附对单层WS2结构和性质的影响。研究结果证实,Fe原子吸附也是在WS2单层中引入磁性相互作用的重要手段。在高覆盖度下Fe/WS2具有半金属性,是一种非常有希望实现的二维半金属材料。
2 计算方法
本文基于密度泛函理论平面波赝势方法,采用VASP (Viennasimulation package)软件进行计算26。以缀加投影波方法(projector-augmented wave method,PAW)27描述离子实和价电子之间的相互作用。应用广义梯度近似(GGA)的PBE方案处理电子间相互作用的交换关联能28。其中平面波截断能量为500 eV。由于Fe原子轨道的强交换作用,在计算过程中考虑了电子的自旋自由度,但没有考虑自旋轨道耦合作用。在几何优化和态密度计算过程中,全Brillouin区积分的点网格取为6 × 6 × 129。结构优化过程中,原子完全弛豫,能量收敛标准为1 × 10−7eV,力收敛标准为0.1 eV∙nm−1。单层原胞选为4 × 4 × 1,每个衬底原胞含有48个原子。为减小层间的相互作用,真空层的厚度取为2.00 nm。优化得到的WS2晶格常数为0.318 nm,与其他理论和实验结果一致30,31。
3 结果与讨论
3.1 Fe原子最稳定吸附位置
为了确定Fe原子在单层 WS2的最稳定吸附位置,根据WS2结构的对称性,我们首先计算了Fe原子在高对称吸附位置的吸附能,其计算公式如下:a=WS2+X−tot,其中tot是Fe-WS2吸附结构的总能,WS2和X分别是清洁单层WS2和孤立Fe原子的总能。如果将原子覆盖度定义为Fe原子和W原子数的比值,在确定稳定位置的计算过程中,Fe原子覆盖度定为0.0625 ML。此时Fe原子的磁性作用很弱,磁性作用(磁序)不会影响最稳定吸附位置的确定。计算过程的磁序是铁磁序,即所有吸附Fe原子的局域磁矩方向相同。单层WS2结构存在3个高对称吸附位置(如图1):W原子的顶位(Tw)、3个S原子与紧邻3个W原子组成的六元环中心的顶位(H)和S原子顶位(Ts)。计算得到不同位置的Fe原子吸附能如表1所示。在Tw位的吸附能最大,约为1.84 eV,比H和Ts位的原子吸附能分别大0.34和1.22 eV。因此Tw位是Fe原子在单层WS2结构的最稳定吸附位置。
在Tw、H和Ts吸附位置,Fe原子与紧邻S原子均成键。Fe―S键键长分别等于0.215、0.211和0.224 nm,数值处于Fe-S系统的Fe―S键键长范围内32。比较3个位置的键长大小可得,在H位结构的Fe与S间作用最强,Tw次之,Ts位最弱。Fe与S原子之间强的化学作用不可避免地会改变了衬底原子间原有的相互作用。在Tw、H和Ts吸附结构中,紧邻吸附位置的WS键键长,分别比清洁WS2衬底的键长增大0.011、0.009和0.001 nm。衬底W与S键变长,说明W―S成键作用减弱。在Tw位Fe原子分别和3个硫原子以及1个W原子近邻,对W―S键的削弱作用最强;而在Ts位Fe原子只与1个S原子近邻,对W―S键的削弱作用最弱。
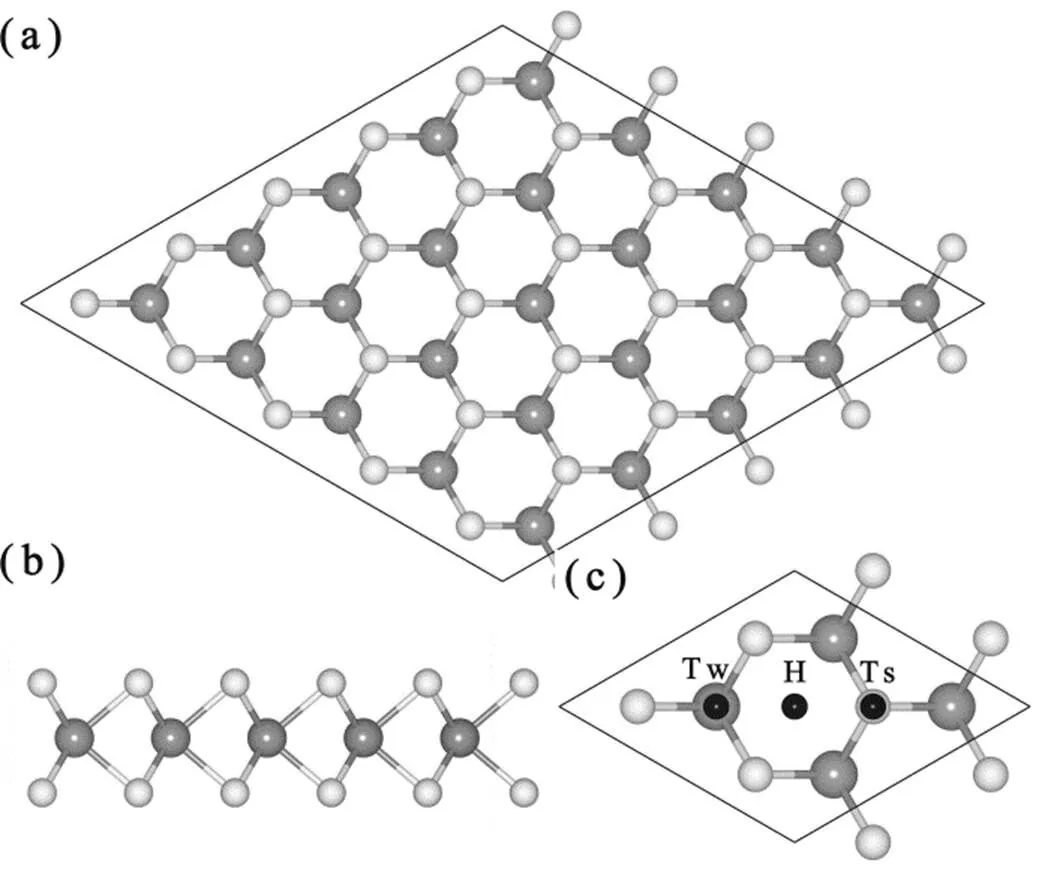
图1 单层WS2的原子结构和Fe原子的可能吸附位置
(a) and (b) are the top and side views of WS2 monolayer. (c) gives the possible adsorption site of Fe atom on WS2 monolayer. The light and gray spheres represent the S and W atoms, respectively. The black spheres indicate possible adsorption sites.
清洁WS2单层没有磁性,Fe原子吸附引入了3轨道的强交换作用,从而使体系表现出一定的磁性。由表1中可得,每个原子引入的磁矩大小和吸附位置有关。在H和Tw位,每个Fe原子引入的局域磁矩约为2.0B,而在Ts位相应的局域磁矩约为4.0B。这说明不同吸附位置的Fe-轨道电子占据数分布不同。引入的磁矩主要集中在Fe原子上。在最稳定的Tw位,Fe、紧邻W和S原子的局域磁矩分别为1.94、0.10和−0.02B。由于W原子轨道巡游性较强,更容易被Fe原子极化,因此W原子的局域磁矩略大于S原子的。Fe原子与紧邻W原子磁矩方向相同,而Fe原子与紧邻S原子是磁矩方向相反。这说明在低覆盖度下Fe原子借助于衬底原子轨道,具有一定成分的超交换作用。
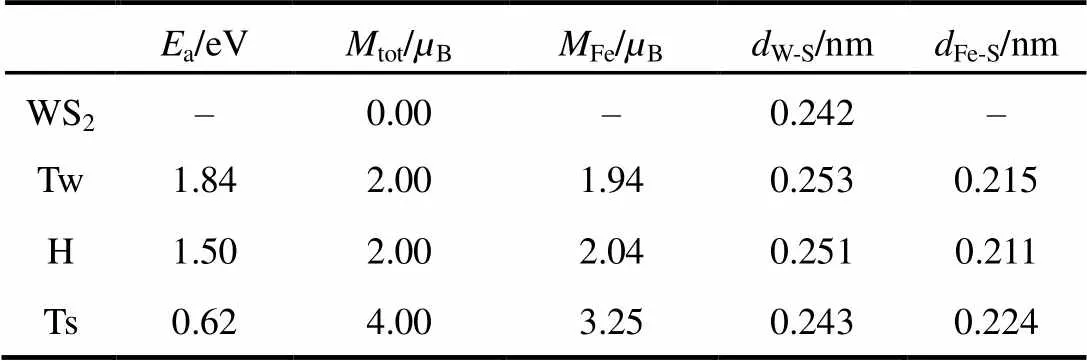
表1 Fe吸附WS2结构的吸附能、原子磁矩和原子间距
a: adsorption energy.A-B: the bond length or nearest distance between A and B.X: atomic magnetic moment of X.
3.2 最稳定吸附结构的电子态
为了进一步说明吸附Fe原子与衬底间的相互作用以及磁性产生的机理,我们计算了清洁WS2和最稳定吸附结构的电子态密度(density of states,DOS),如图2所示。在单层WS2结构中,每个S原子(3234)(此类用斜体)从近邻的W原子(5462)获得2个电子。S原子的3轨道全占据,呈−2价;W原子的5轨道局域占据(52),呈+4价。根据泡利不相容和能量最低原理,W原子最外层剩余的两个电子以自旋反平行的形式占据能量最低轨道。清洁的单层WS2的电子态是自旋向上与向下对称分布,非自旋极化的,如图2(a)所示。在最稳定吸附结构中,Fe原子的吸附引入了磁性,使自旋向上和向下的电子态呈非对称分布,在费米能附近出现了Fe原子局域轨道形成的杂质峰(如图2(b)所示)。根据原子局域磁矩的计算结果,磁性主要集中在Fe原子上。最稳定的Tw吸附位置具有3对称性,因此Fe原子的3轨道劈裂为3个简并能级:d,x2–y2、d2和d,xz(如图2(d)所示)。在自旋向上通道,所有轨道全占据;在自旋向下通道,d,x2−y2和d2轨道被电子占据,而d,xz轨道全空。Fe原子的最外层价电子占据数约等于8,与孤立Fe原子3轨道电子数相等。这说明吸附Fe原子和衬底WS2间没有明显的电荷转移,吸附作用主要使Fe-轨道的电子发生的重新分布。不同简并轨道的自旋劈裂不同。d,x2−y2和d2自旋劈裂能分别约为−0.01和−0.22 eV,这主要是由轨道空间分布的差异导致不同的库仑作用和自旋交换作用引起的。由于Fe原子的吸附作用,紧邻W和S原子在费米能附近出现了和Fe原子局域轨道一致的杂化峰。在图2(c)中Fe与W原子的轨道杂化峰强度明显强于与S原子的。这说明Fe和W原子轨道存在较大空间的交叠(Overlap),两者间轨道杂化作用较强。这与Tw位Fe原子的吸附能比H位大的结果一致。由于轨道的方向性,Fe原子d2轨道和W轨道的杂化作用最弱,而与d,x2−y2轨道杂化作用最强。
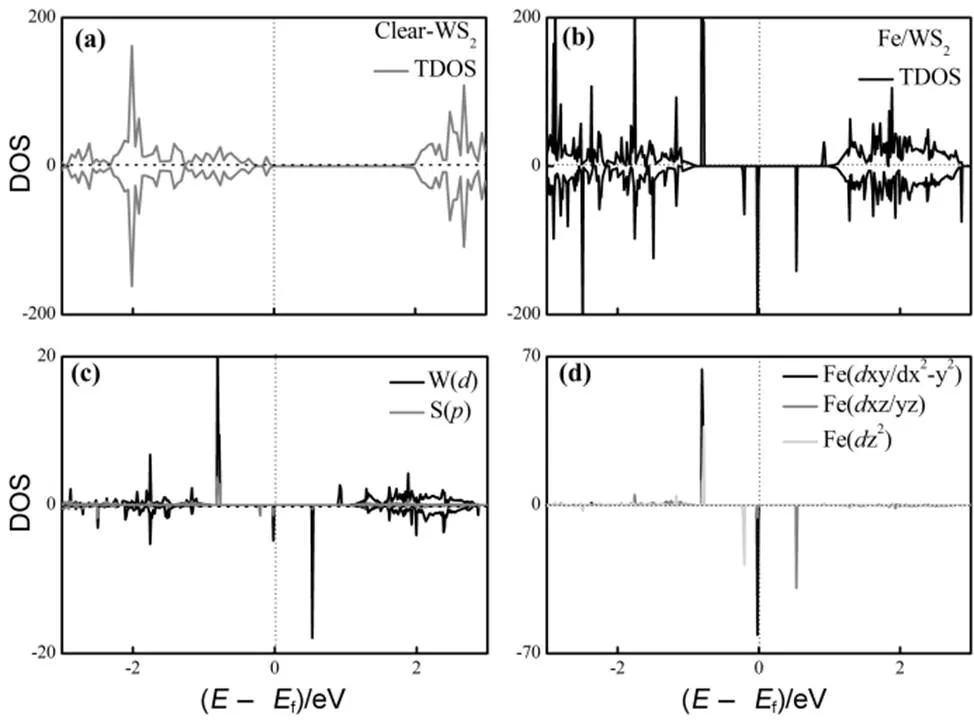
图2 清洁单层WS2和Fe/WS2的总电子态密度和投影电子态密度
(a) and (b) are the total electron density of states (DOS) of the clear WS2 and the Fe/WS2, respectively; (c) and (d) are the projected density of states (PDOS) of the nearest W, S and the Fe atoms, respectively.
为了说明吸附作用引起的吸附位置附近电荷分布的变化,我们计算了吸附结构的电荷差分密度,如图3所示。电荷差分密度计算公式为
Δ=Fe/WS2−WS2−Fe
其中,Fe/WS2、WS2和Fe分别是吸附结构、清洁衬底和孤立Fe原子的电荷密度。在计算3个体系的电荷密度过程中,原胞相同,相应的原子位置相同。由图3可得,电荷分布的变化主要集中在Fe原子周围。Fe原子上方的电荷减少,而下方靠近衬底的区域电荷增加。这说明Fe和衬底间存在较强的相互作用。在衬底中,紧邻W―S键附近的电荷密度减小。这说明Fe原子的吸附作用削弱了W―S键。这与W―S键长变化趋势相吻合。电荷差分密度也说明Fe和衬底间不存在明显的电荷转移,而电荷转移主要发生在Fe原子不同轨道之间。这与PDOS和局域磁矩的计算结果一致。
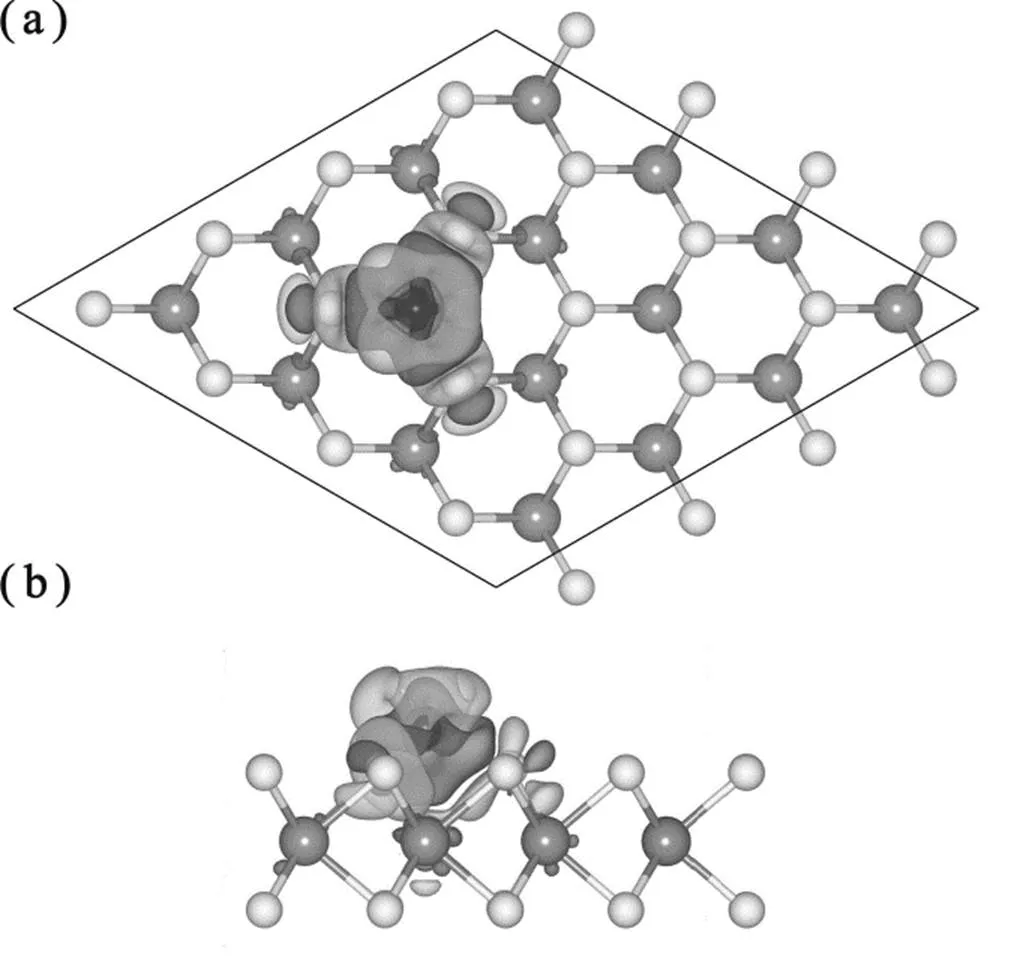
图3 Fe-WS2结构电荷差分密度图
Dark (light) surface represents aera of getting (losing) electron,the-value is set to be ±11∙nm−3.
3.3 不同覆盖度下的磁相互作用和性质
在低覆盖度下,虽然Fe原子吸附可以在单层WS2引入磁性。为了形成不同的自旋通道,系统必须能够形成稳定的铁磁序。在低覆盖度下,Fe原子间的作用以超交换作用为主,铁磁序是不稳定的。为了寻找具有稳定铁磁序的结构,我们讨论Fe/WS2结构在不同覆盖度下的基态磁序。Fe/WS2体系具有3对称性,并不存在严格意义的反铁磁结构。为了讨论Fe/WS2体系的铁磁序,我们计算了图4所示的铁磁和反铁磁序的能= (AFM−FM)/,其中AFM和FM分别是反铁磁序与铁磁序的结构总能,是原胞中的Fe原子数目。能差> 0说明铁磁序更稳定,反之则铁磁序不稳定。在计算过程中我们选取了4种覆盖度:0.125、0.25、0.5和1.0 ML。计算过程均采用4 × 4 × 1的超原胞。计算结果表明,0.125和0.25 ML低覆盖度的能差小于零,分别为−21.9和−9.81 meV每个Fe原子。这说明在低覆盖度下铁磁序不稳定。而0.5和1.0 ML覆盖度的能差大于零,分别等于175和174 meV每个Fe原子。这说明在高覆盖度下铁磁序更稳定。
为了说明相互作用随覆盖度的变化,我们计算了四种覆盖度下的原子间距。如表2所示,首先在0.125–1.0 ML四种覆盖度下,Fe原子最近距离分别为1.102、0.636、0.435和0.318 nm。随着覆盖度增加,Fe原子间距逐渐减小,Fe原子间相互作用增强。Fe原子与紧邻W原子的距离由0.125 ML覆盖度下的0.255 nm逐渐增大至1.0 ML覆盖度下的0.282 nm。而Fe原子与紧邻S原子的距离随着覆盖度的增加略微增大,但变化趋势并不明显。紧邻W与S原子键长随覆盖度的增加而逐渐减小。这些原子间距的变化说明,随着覆盖度的增加,Fe原子与衬底间的相互作用也随之逐渐减弱。
为了估算原子间的相互作用能,我们也计算了不同覆盖度下Fe原子在Tw和H位的原子吸附能,如表2所示。低覆盖度(0.125和0.25 ML)的原子吸附能和0.0625 ML的非常接近,两者的差值小于0.02 eV。这是由于在低覆盖度下吸附Fe原子之间的距离较大(大于0.636 nm),Fe原子间的直接相互作用很弱造成的。因此我们认为在最稳定吸附位置,Fe原子和衬底硫原子的相互作用能约为1.49 eV(H位的吸附能)。而Fe原子和紧邻W原子的作用能约为0.34 eV (为Tw和H吸附能的差值)。随着覆盖度增加,Fe原子间的间距减小,相互作用能增加。Fe原子间的直接作用在0.5和1.0 ML覆盖度下约为0.29和0.54 eV。
为了说明磁性相互作用与覆盖度的关系,我们也计算了Fe原子和紧邻原子间的局域原子磁矩(如表2所示)。由表可得,在同一吸附结构中,Fe原子磁矩最大,其数值远远大于紧邻W和S原子的磁矩,其中S原子的磁矩最小。这是因为吸附原子轨道电子具有较强的局域性,易受库仑作用和自旋交换作用影响,因此磁矩主要集中在Fe原子上。由于W原子轨道电子的巡游性,易被Fe原子极化,在同一种体系中,W原子的局域磁矩大于S原子的。在低覆盖度下,Fe与紧邻W和S原子磁矩方向分别相同和相反,说明Fe与紧邻W和S原子分别为铁磁耦合和反铁磁耦合作用。这是说明在低覆盖度下Fe原子存在超交换作用。在高覆盖度下,Fe原子和衬底的磁性作用发生了明显的变化。Fe与W原子磁矩方向相反,与S原子磁矩方向相同。这主要是因为Fe原子间的相互作用导致轨道对应的能级宽度增加,费米能附近的巡游电子和Fe原子的局域磁矩存在RKKY相互作用导致的33–35。

图4 在0.125 ML (a),0.25 ML (b),0.5 ML (c)和1.0 ML (d)覆盖度下的铁磁和反铁磁序结构
The black spheres represent the iron atoms and the arrows represent the direction of the electronic spin.

表2 不同覆盖度下的Fe原子吸附能、磁序能差、磁矩以及原子间距
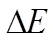
为了从电子态上说明相互作用随覆盖度的变化,我们也计算了覆盖度从0.125至1.0 ML范围內Fe原子轨道的投影电子态密度(projected density of states,PDOS)。在低覆盖度下,Fe原子轨道电子态是一些比较窄的杂化峰。Fe原子的3轨道劈裂为3个简并能级:d,x2−y2、d2和d,yz,如图5(a)和(b)所示。在自旋向上通道,轨道全占据,轨道能量均位于费米能以下−1.0 eV附近。在自旋向下通道,占据轨道能量位置有所不同,分别位于−0.21和−0.02 eV附近。空轨道能级在0.53 eV附近。在高覆盖度下,由于吸附原子间的相互作用增强,对应轨道的电子态宽度增加。在自旋向上通道,轨道能量位于−2.5 – −1.5 eV范围內;在自旋向下通道,占据轨道能量在−1.0 – −0.4 eV范围,空轨道能量位于0.5–0.7 eV区间。从轨道能量变化角度上看,随着覆盖度的增加,d,yz轨道得到电子,而d2轨道失去电子。这说明在自旋向下通道中部分电子从d2向d,yz轨道转移。这主要是在高覆盖度下Fe原子间相互作用增强引起的,同时在一定程度上削弱了Fe和衬底的相互作用。
为了说明高覆盖度下铁磁序吸附体系的性质,我们分别计算了0.5和1.0 ML覆盖度下吸附结构的总电子态密度(TDOS),如图6(a, b)所示。由图可得,在0.5和1.0 ML覆盖度下,费米能级穿过自旋向下通道的电子态,而在自旋向上通道,费米能级及其附近没有明显的电子态。费米能级附近的电子极化率为100%。这说明该吸附结构具有半金属性。此外我们也计算了1.0 ML覆盖度下的能带结构,如图7所示。在自旋向下通道,价带顶和导带底分别位于布里渊区的和点,形成间接带隙。能隙宽度约为0.94 eV,比清洁单层WS2的禁带宽度(~1.82 eV)小约0.88 eV。这说明在高覆盖度下Fe原子吸附改变了单层WS2的半导体性质,使其具有半金属性质。

图5 在0.125 ML (a),0.25 ML (b),0.5 ML (c)和1.0 ML (d)覆盖度下的Fe原子d轨道的投影态密度
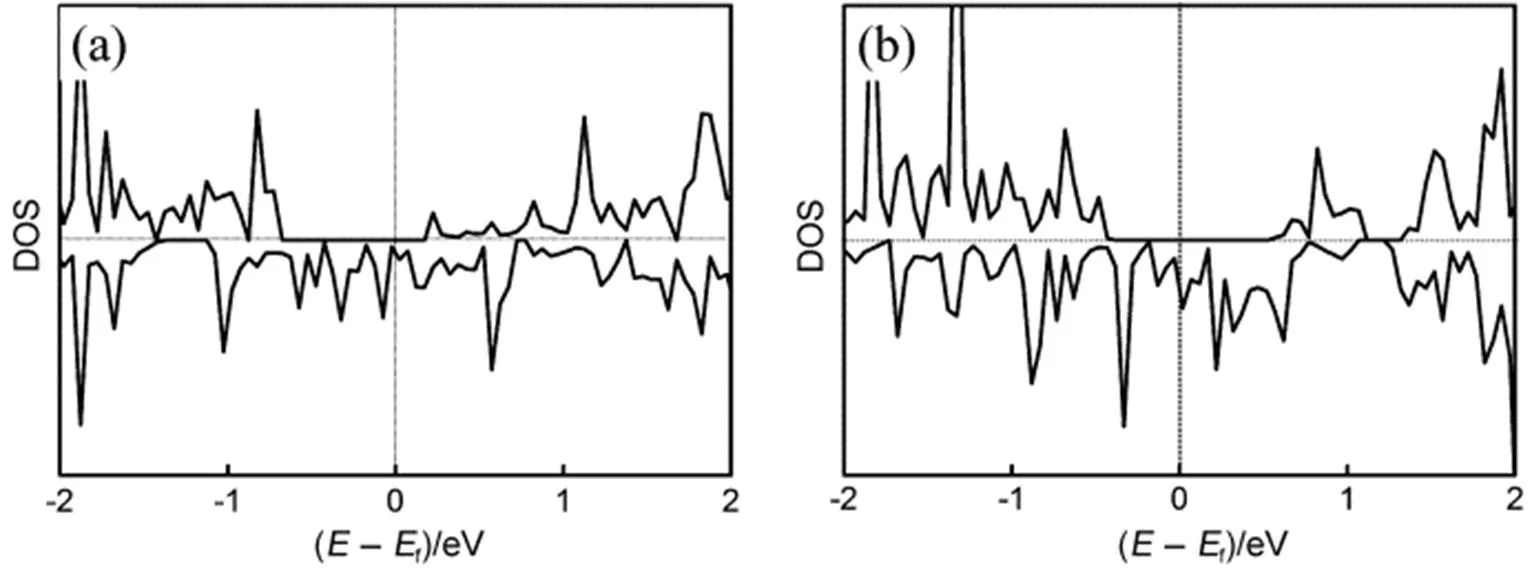
图6 在0.5 ML (a)到1.0 ML (b)覆盖度下Fe/WS2结构的总态密度
图7 在1.0 ML覆盖度下Fe/WS2结构自旋向上(a)和自旋向下通道(b)的能带结构
4 结论
我们采用以密度泛函理论为基础的第一性原理方法研究了Fe原子吸附对单层WS2结构和性质的影响。研究结果表明,W原子的顶位是Fe原子在WS2单层的最稳定吸附位置,相应的原子吸附能最大,约为1.84 eV。Fe与衬底原子间的轨道杂化作用,削弱了紧邻W与S原子的成键作用,使相应键长增大0.011 nm。受衬底原子影响,Fe (3642)原子轨道缺失自旋向下的电子,局域磁矩约为2.0B。在低覆盖度下(0.125、0.25 ML),吸附原子间距较大,磁性作用以超交换作用为主,铁磁序不稳定。在高覆盖度下(0.5和1.0 ML),吸附原子间距缩小,磁性作用以RKKY作用为主,铁磁序更稳定。随着覆盖度增加,Fe原子间相互作用增强,Fe–Fe间距由1.102 nm减小至0.318 nm。Fe与衬底原子间的相互作用也随着覆盖度增加而减弱。Fe–W (Fe–S)最紧邻间距随着覆盖度增加,由0.255 (0.214) nm增大至0.282 (0.218) nm。电子态密度的计算结果表明,在0.5和1.0 ML覆盖度下,吸附体系费米能级处的电子极化率等于100%,显示半金属性。自旋向上与向下通道分别为间接带隙的半导体和金属。1 ML覆盖度吸附体系的禁带宽度高达0.94 eV。这说明在高覆盖下,Fe原子吸附可以将直接带隙的WS2半导体转变成半金属,是一种潜在的自旋电子器件材料。
(1) Wang, Q. H.; Kourosh, K. Z.; Kis, A.; Coleman, J. N.; Strano, M. S.. 2012,, 699. doi: 10.1038/nnano.2012.193
(2) Butler, S. Z.; Hollen, S. M.; Cao, L.Y.;.2013,, 2898.doi: 10.1021/nn400280c
(3) Yue, Q.; Shao, Z. Z.; Chang, S. L.; Li, J. B.2013,, 425.doi: 10.1186/1556-276X-8-425
(4) Chhowalla, M.; Shin, H. S.; Eda, G.; Li, L. J.; Loh, K. P.; Zhang, H.2013,, 263. doi: 10.1038/nchem.1589
(5) Haldar, S.; Vovusha, H.; Yadav, M. K.; Eriksson, O.; Sanyal, B.2015,,235408. doi: 10.1103/PhysRevB.92.235408
(6) Liu, H. S.; Han, N. N.; Zhao, J. J.2015,, 17572. doi: 10.1039/C4RA17320A
(7) Yazyev, O. V.; Kis, A.2015,, 20. doi: 10.1016/j.mattod.2014.07.005
(8) Wolf, S. A.; Awschalom, D. D.; Buhrman, R. A.; Daughton, J. M.; von Molnar, S.; Roukes, M. L.; Chtchelkanova, A. Y.; Treger, D. M.2001,, 1488. doi: 10.1126/science.1065389
(9) Cuticle, I.; Fabian, J.and Sarma, S. D.2004,, 323. doi: 10.1103/RevModPhys.76.323
(10) Felser, C.; Fecher, G. H.; Balke, B.2007,, 668. doi: 10.1002/anie.200601815
(11) Zhu, H. J.; Ramsteiner, M.; Kostial, H.; Wassermeier, W.; Schönherr H.P.; Ploog, K, H.2001,, 016601. doi: 10.1103/PhysRevLett.87.016601
(12) Sun, H.; Li, B.; Zhao, J.2016,, 42. doi: 10.1088/0953-8984/28/42/425301
(13) de Groot, R. A.; Mueller, F. M.; van Engen, P. G.; Buschow, K. H. J.1983,, 2024. doi: 10.1103/PhysRevLett.50.2024
(14) Nanda, B. R. K.; Dasgupta, I.2003,, 7307. doi: 10.1088/0953-8984/17/33/008
(15) Lewis, S.P.; Allen, P. B.; Sasaki, T.1997,, 10253. doi: 10.1103/PhysRevB.55.10253
(16) Jedema, F. J.; Filip, A.T.; van Wees, B. J.2001,, 345. doi: 10.1038/35066533
(17) Szotek, Z.; Temmerman, W. M.; Svane, A.; Petit, L.; Winter, H.2003,, 104411. doi: 10.1103/PhysRevB.68.104411
(18) Liu, J.; Zhang, B. Chen, L.; Chen, P. D.; Dong, H. N.; Zheng, R. L.. 2011,, 2101. [刘 俊, 张 博, 陈 立, 陈培达, 董会宁, 郑瑞伦. 物理化学学报, 2011,, 2101.] doi: 10.3866/PKU.WHXB20110724
(19) Liu, J.; Liu, Y.; Chen, X. M.; Dong, H.N.2009,, 107. [刘 俊, 刘 宇, 陈希明, 董会宁. 物理化学学报, 2009,, 107.] doi: 10.3866/PKU.WHXB20090119
(20) Kronik, L.; Jain, M.; Chelikowsky, J. R.2002,, 041203(R). doi: 10.1103/PhysRevB.66.041203
(21) Hong, J.; Wu. R. Q.2005,, 063911. doi: 10.1063/1.1857052
(22) Picozzi, S.; Shishidou, T.; Freeman, A. J.; Delley, B.2003,, 165203. doi: 10.1103/PhysRevB.67.165203
(23) Ramasubramaniam, A.; Naveh,D.2013,,195201. doi: 10.1103/PhysRevB.87.195201
(24) Li, H. P.; Liu, S.; Huang, S. L.; Yin, D. Q.; Li, C. S.; Wang, Z. C.2016,, 2364. doi: 10.1016/j.ceramint.2015.10.033
(25) Zhao, X.; Dai, X. Q.; Xia, C. X.; Wang, T. X.2015,, 339. doi: 10.1016/j.spmi.2015.06.007
(26) Kresse, G.; Furthmuller, J.1996,, 11169. doi: 10.1103/PhysRevB.54.11169
(27) Kresse, G.; Joubert, J.1999,, 1758. doi: 10.1103/PhysRevB.59.1758
(28) Perdew, J. P.; Burke, K.; Ernzerhof, M.1996,, 3865. doi: 10.1103/PhysRevLett.77.3865
(29) Monkhorst, H. J.; Pack, J. F.1979,, 5188. doi: 10.1103/PhysRevB.13.5188
(30) Liu, H. S.; Han, N. N.; Ji, J.2015,, 17572. doi: 10.1039/C4RA17320A
(31) Haldar, S.; Vovusha, H.; Yadav, M. K.; Eriksson, O.; Sanyal, B.2015,. 235408. doi: 10.1103/PhysRevB.92.235408
(32) Lewis, S. R.1925,10, 281-304.doi: http://rruff.info/doclib/am/vol10/AM10_281.pdf
(33) Schwabe, N. F.; Elliott, R. J.1996,, 12953. doi: 10.1103/PhysRevB.54.12953 Litvinov, V. I.; Dugaev, V. K.1998,, 3584. doi: 10.1103/PhysRevB.58.3584
(34) Ziener, C. H.; Glutsch S.; Bechstedt, F.2004,, 075205. doi: 10.1103/PhysRevB.70.075205
Effect of Adsorption of Fe Atoms on the Structure and Properties of WS2Monolayer
XU Wei-Yun1,2WANG Li-Li1MI Yi-Ming1ZHAO Xin-Xin1,*
(1;2)
In this work, first-principles calculations were performed to study the effect of the adsorption of Fe atoms on the structure and properties of the WS2monolayer. It was found that the most stable adsorption site for an Fe atom on WS2at low coverage (<0.0625 ML) of the monolayer lies directly above the W atom and the atomic adsorption energy is ca. 1.84 eV. The interaction between the Fe and substrate atoms weakens the nearest W―S bonds which increases their bond length by ca. 0.011 nm. The orbital occupation of the adsorbed Fe atoms also undergoes redistribution. The 3orbitals of Fe are fully occupied with the exception of the spin down channel ofdanddorbitals. The magnetic interactions of Fe−Fe are mainly believed to involve super-exchange interactions which are mediated by the substrate. Thus the ferromagnetic order is unstable at low coverage. However, at high coverage, the distance between Fe−Fe decreases and the states close to the Fermi energy level induce magnetic interactions between the local magnetic moment and the itinerant electron, which are identified as RKKY interactions. In this manner, the ferromagnetic order is more stable at high coverage of the monolayer. The Density of state and band-structure calculations show that the spin polarization of Fe−WS2near the Fermi energy level is about 100%. The spin up channel acts as an indirect band gap semiconductor, while the another one acts as a metal. These calculations indicate that the Fe-WS2layer at high coverage could be half metallic, which can be potentially used to develop spin-based electronic materials.
DFT; Fe/WS2; Transition metal dichalcogenides; Exchange interaction; Half-metal
March 24, 2017;
April 28, 2017;
May 10, 2017.
. Email: bighunter@sues.edu.cn; Tel: +86-21-67791191.
10.3866/PKU.WHXB201705102
O641
The project was supported by the National Natural Science Foundation of China (11504228).
国家自然科学基金(11504228)资助项目
猜你喜欢
杂志排行

物理化学学报的其它文章
- ZAβ3和Aβ16–40亲和作用的分子机理解析
- 选择氧化腈化C―H键制备有机腈类化合物
- Co4O4立方烷分子催化剂用于高效光电催化分解水
- 高效钙钛矿缺陷态钝化材料及其钝化机理
- Design of Benzobisthiadiazole Analogues as Promising Anchoring Groups for High Efficient Dye-Sensitized Solar Cells
- Theoretical Studies on the Structures and Opto-Electronic Properties of Fluorene-Based Strained Semiconductors