查尔酮衍生物抗肿瘤作用机制及其构效关系研究进展
2017-12-20饶国武浙江工业大学药学院浙江杭州310014
刘 岩,饶国武(浙江工业大学药学院,浙江 杭州 310014)
查尔酮衍生物抗肿瘤作用机制及其构效关系研究进展
刘 岩,饶国武*
(浙江工业大学药学院,浙江 杭州 310014)
查尔酮化合物由于结构简单、易于合成且具有抗肿瘤活性而得到广泛研究。本文归纳总结了查尔酮化合物的抗肿瘤机制及其构效关系,为其结构修饰提供了理论基础。
查尔酮;抗肿瘤;作用机制;构效关系
0 前言
癌症已经成为威胁人类生命健康的常见病、多发病。随着分子肿瘤学,分子生物学的不断发展,以及纳米技术在药物中的不断应用,高效低毒的分子靶向抗肿瘤药物成为药物学家研究的热点。查尔酮作为黄酮化合物的重要前体,已经报道其具有非常广泛的生理活性,特别是在抗肿瘤方面已经取得了初步进展。本文主要对查尔酮的抗肿瘤机制及其构效关系进行了总结。
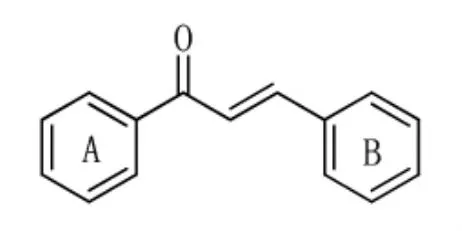
Figure1 查尔酮骨架
1 查尔酮的抗肿瘤作用机制
1.1 抗血管生成
血管生成在肿瘤细胞的生长和代谢中起着非常关键的作用,新血管可以为肿瘤细胞和组织的生长提供充足的氧气和营养物质,因此,以VEGF/VEGFR作为靶点的抗肿瘤药物成为临床试验中癌症治疗的一个关键策略。
王治愈等[1]从甘草根中提取出了异甘草素(化合物1,Figure 2),体外毛细管形成试验证明化合物1可以抑制血管内表皮生长因子诱导的细胞增殖。并通过鸡主动脉环模型进一步证实了其抗血管生成活性。同时研究了其抑制机理。结果表明,化合物1可以通过促进缺氧诱导因子蛋白酶体的降解和抑制VEGFR-2信号通路来抑制血管内表皮生长因子的表达,以此抑制肿瘤细胞的生长。
L.Varinska, S.Emami等[2]使用苯乙酮或环酮与取代苯甲醛,在氢氧化钠或哌啶的催化下合成了一系列查尔酮或环状的查尔酮类似物(Scheme 1)。通过血管生成模型测试了化合物2(Figure 2)的抗血管生成活性,展现了显著的抑制活性。机理研究证明化合物2通过抑制b-FGF和VEGF诱导的AKT和ERK的磷酸化来抑制血管生成。
Ku等[3]研究了查尔酮衍生物4’-乙酰氨基-4-羟基查尔酮(化合物3,Figure 2)的抗肿瘤活性及机理。通过体外的人脐静脉内皮细胞测试了其抗血管生成活性。发现化合物3可以显著抑制VEGF诱导的人脐静脉内皮细胞的迁移和入侵。

Figure 2 查尔酮化合物的结构

Scheme 1
1.2 抑制细胞周期
细胞的生长分裂必须经过完整的细胞周期,细胞周期的运行受控于精密的细胞周期调控机制,其中细胞周期蛋白依赖性激酶(CDK)是调控系统的核心蛋白酶,还包括CDC25B,aurora等激酶。细胞癌变过程中,通常伴随着CDK活性失去控制,细胞周期会处于失控状态。由于微管蛋白参与有丝分裂过程纺锤体的形成,故可以通过抑制微管蛋白来抑制细胞的有丝分裂过程,从而产生抗肿瘤活性。
胡永洲等[4]以夫拉平度(Flavopiridol,Figure 2)为先导化合物,设计合成了化合物4(Figure 2),以间苯三酚为起始原料,通过傅克酰基化,烷基化合成取代苯乙酮,再与取代苯甲醛缩合生成目标化合物(Scheme 2)。体外CDK/Cyclic B抑制活性试验表明,多数衍生物对CDK/Cyclic B具有强效的抑制活性。李艳玲等[5]合成了一系列类黄酮类化合物(化合物5,Figure 2),体外 HCT116抑制 IC50均在 2~5 μmol/L。 当 R=p-Cl,p-OCH3,p-Br时,体外CDK1抑制活性均高于参照药物夫拉平度。

Scheme 2
Yuan等[6]研究了甘草查尔酮(化合物6,Figure 2)的抗肿瘤作用机理。体外和体内的抗恶性膀胱癌细胞株试验表明化合物6可以明显抑制细胞株增殖,使细胞株细胞分裂期的S期停滞,减少cyclinA和CDK1、CDK2,CDC25A/B蛋白的表达。
Aurora激酶是丝氨酸/苏氨酸激酶家族的新成员,在很多肿瘤细胞中过表达,在细胞周期的控制中起着重要作用。Shin等[7]合成了一系列苯并吡喃查尔酮衍生物 (Scheme 3),体外HCT116结直肠癌细胞株存活实验表明化合物7(Figure 2)的IC50可达到93.1 nmol/L。体外的结合试验发现此化合物可以抑制Aurora激酶。
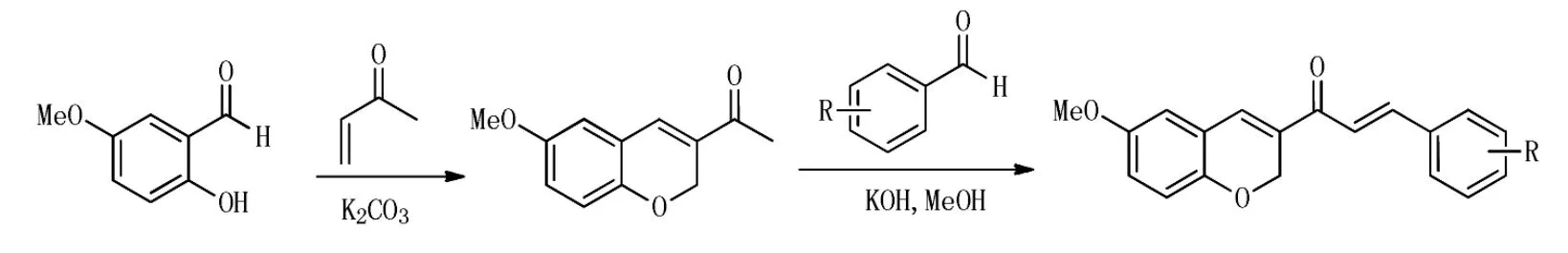
Scheme 3
文献报道[8-10],吲哚衍生查尔酮化合物8(R=H)可以选择性抑制膀胱癌细胞的增殖,分子对接结果表明,化合物8在秋水仙碱结合部位与微管蛋白有较强相互作用。体外抑制肿瘤细胞增殖试验表明化合物9(R=OCH3)可以通过使细胞周期的S期停滞来抑制细胞增殖。

Scheme 4
1.3 阻碍信号通路
细胞生长不仅与细胞周期的相关激酶的控制有关,而且还受到细胞内外信号转导系统的调控。生长因子等胞外信号与受体结合产生的刺激通过信号通路传递到细胞内,从而调控细胞的增殖、分化等进程。其中,最重要的三条通路为:由磷酯酰肌醇3-激酶(PI3K)和其下游的蛋白激酶B(PKB/Akt)、雷帕霉素靶体蛋白(mTOR)组成的PI3K-AKT-mTOR通路;丝苏氨酸蛋白激酶Ras和丝裂原活化蛋白激酶(MAPK)三级级联激酶组成的Ras-MAPK通路;以及下游信号转导与转录激活因子STAT家族。
Sun等[11]报道了甲氧基查尔酮衍生物(化合物10,Figure 2)可以阻碍蛋白质的合成,抑制mTOR信号传导通路。该化合物对mTOR通路的抑制主要归因于线粒体应激下调蛋白质存活水平。
BRAF蛋白是RAS/MAPK信号通路的一部分,其调控着细胞的增殖和分化。突变型BRAF基因允许BRAF单独上调下游信号,通过MEK和ERK使下游信号高度活化,反过来导致细胞的过度增殖和存活。Li等设计并合成了一系列的(E)-α-苯磺酰基查尔酮衍生物 (合成路线见Scheme5)。化合物 11(Figure1)显示了有潜力的抑制活性,对B-RAFV600E的IC50值达到0.17μmol/L。

Scheme 5
JAK/STAT信号级联介导许多重要的细胞功能,如细胞增殖,凋亡和免疫应答等。这条通路的突然激活会导致恶性肿瘤的转化和异常生长。通过减少此通道的异常磷酸化可以抑制肿瘤细胞的生长[12-13]。 Pinz等[14]报道了 α-溴-2,3’,4,4’-三甲氧基查尔酮(化合物12,Figure 2)可以通过抑制JAK2和STAT5的磷酸化来抑制JAK/STAT信号通路。染色质免疫沉淀反应试验证明该化合物可以减少RNA聚合酶Ⅱ与STAT5靶向基因的结合。
1.4 其他作用机制
查尔酮及其衍生物除上述抗肿瘤作用机制外,还具有广泛的细胞毒性[15-16],诱导细胞凋亡[17-18],抗多药耐药性[19]等作用机制。
2 构效关系研究
由于查尔酮结构具有广泛的药理活性,故化学家在保持查尔酮基本骨架的同时,对丙烯酮连接的两个苯环进行结构修饰。并通过体外或体内的活性筛选得到了许多具有较好抗肿瘤活性的化合物。以夫拉平度作为先导化合物进行结构改造时发现,哌啶环被一个连有亚甲基的胺基环代替后同样得到较好的CDK抑制活性,且B环的对位引入卤素、甲氧基等基团时,化合物同样具有较好的抑制活性。
吲哚是一个优良的药效基团,将查尔酮的B环替换为吲哚环后发现该化合物可以显著抑制微管蛋白聚合,2,4位的甲氧基同样具有重要作用,6位的甲氧基不是必需基团。当在A环引入多甲氧基基团结构,化合物在JAK/STAT通路中有着显著的抑制作用。此外,对丙烯酮结构的修饰同样可以影响化合物的抗肿瘤活性。当将亚砜基接入碳碳双键,发现该化合物对BRAF细胞具有明显的抑制作用;当双键氢被甲氧基或氨基取代时,化合物可以明显减弱细胞的有丝分裂,抑制细胞的增殖。
3 总结
随着分子生物学和肿瘤科学的不断发展,肿瘤发生的作用机制逐渐被揭开,促进了分子靶向药物的发展。查尔酮及其衍生物由于结构简单,合成难度较小,且具有广泛的抗肿瘤活性及其多种抗肿瘤机制,成为抗肿瘤药物研发过程中的重要研究方向。本综述对查尔酮的作用机制及构效关系做了简要介绍,为以后查尔酮的结构修饰和改造提供理论依据。
[1] Bertl E,Becker H,Eicher T,et al.Inhibition of endothelial cell functions by novel potential cancer chemopreventive agents[J].Biochemical and biophysical research communications, 2004,325(1):287-295.
[2] Varinska L,Van Wijhe M,Belleri M,et al.Anti-angio-genic activity of the flavonoid precursor 4-hydroxychalcone[J].European journal of pharmacology, 2012, 691 (1):125-133.
[3]Ku B M,Ryu H W,Lee Y K,et al.4′-Acetoamido-4-hydroxychalcone, a chalcone derivative, inhibits glioma growth and invasion through regulation of the tropomyosin 1 gene[J].Biochemical and biophysical research communications, 2010, 402(3):525-530.
[4] 杜立林.含氮查尔酮衍生物CDK1抑制剂的设计、合成及构效关系研究[D].杭州:浙江大学,2010.
[5] 李艳玲,方浩,徐文芳 类黄酮细胞周期蛋白依赖性激酶抑制剂的合成与抗癌活性研究[J].包头医学院学报,2011, 27(4) :12-14.
[6]Yuan X, Li T, Xiao E, et al.Licochalcone B inhibits growth of bladder cancer cells by arresting cell cycle progression and inducing apoptosis[J].Food and Chemical Toxicology, 2014, 65:242-251.
[7]Shin S Y,Yoon H,Ahn S,et al.Chromenylchalcones showing cytotoxicity on human colon cancer cell lines and in silico docking with aurora kinases[J].Bioorganic&medicinal chemistry, 2013, 21(14):4250-4258.
[8] Boumendjel A,McLeer-Florin A,Champelovier P,et al.A novel chalcone derivative which acts as a microtubule depolymerising agent and an inhibitor of P-gp and BCRP in in-vitro and in-vivo glioblastoma models[J].BMC cancer, 2009, 9(1):242.
[9] Martel-Frachet V,Areguian J,Blanc M,et al.Investigation of a new 1,3-diarylpropenone as a potential antimitotic agent targeting bladder carcinoma[J].Anti-cancer drugs, 2009, 20(6):469-476.
[10]Martel-Frachet V,Keramidas M,Nurisso A,et al.IPP51,a chalcone acting as a microtubule inhibitor with in vivo antitumor activity against bladder carcinoma[J].Oncotarget, 2015, 6(16):14669-14686.
[11]Sun Y W,Huang W J,Hsiao C J,et al.Methoxychalcone induces cell-cycle arrest and apoptosis in human hormone-resistant prostate cancer cells through PI 3-kinaseindependent inhibition of mTOR pathways[J].The Prostate,2010, 70(12):1295-1306.
[12]Harrison D A.The jak/stat pathway[J].Cold Spring Harbor perspectives in biology, 2012, 4(3):a011205.
[13]Zhang Q,Raje V,Yakovlev V A,et al.Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727[J].Journal of Biological Chemistry,2013, 288(43):31280-31288.
[14]Pinz S, Unser S, Brueggemann S, et al.The synthetic αbromo-2′, 3, 4, 4′-tetramethoxychalcone (α-Br-TMC)inhibits the JAK/STAT signaling pathway[J].PloS one,2014, 9(3):e90275.
[15]Kamal A,Ramakrishna G,Raju P,et al.Synthesis and anti-cancer activity of chalcone linked imidazolones[J].Bioorganic&medicinal chemistry letters, 2010, 20(16):4865-4869.
[16]Miranda C L,Stevens J F,Helmrich A,et al.Antiproliferative and cytotoxic effects of prenylated flavonoids from hops (Humulus lupulus) in human cancer cell lines[J].Food and Chemical Toxicology, 1999, 37(4):271-285.
[17]Fang X,Yang B,Cheng Z,et al.Synthesis and antitumor activity of novel nitrogen mustard-linked chalcones[J].Archiv der Pharmazie, 2013, 346(4):292-299.
[18]Shenvi S, Kumar K,Hatti K S,et al.Synthesis, anticancer and antioxidant activities of 2, 4, 5-trimethoxy chalcones and analogues from asaronaldehyde:Structureactivity relationship[J].European journalofmedicinal chemistry, 2013, 62:435-442.
[19]Winter E, Gozzi G J, Chiaradia-Delatorre L D,et al.Quinoxaline-substituted chalcones as new inhibitors of breast cancer resistance protein ABCG2:polyspecificity at B-ring position[J].Drug design,development and therapy,2014, 8:609.
Advances in Anti-tumor Mechanism and Structure-activity Relationship of Chalcone Derivatives
LIU Yan,RAO Guo-wu*
(College of Pharmaceutical Science,Zhejiang University of Technology,Hangzhou,Zhejiang 310014, China)
Chalcone compounds are extensively studied due to their simplicity, ease of synthesis and extensive antitumor activity.This thesis summarized the anti-tumor mechanism and structure-activity relationship of chalcone compounds,and provided the theoretical basis for the subsequent structural modification.
chalcone;antitumor;mechanism;structure-activity relationship
1006-4184(2017)11-0015-05
2017-04-09
刘岩(1990-),男,山东泰安人,硕士研究生在读,主要从事药物分子设计与合成研究。*
饶国武,E-mail: rgw@zjut.edu.cn。
