吲哚在FAU分子筛中吸附行为的分子模拟研究
2017-12-06刘熠斌
党 宇,丁 雪,刘熠斌,冯 翔
(中国石油大学(华东)重质油国家重点实验室,山东 青岛 266580)
吲哚在FAU分子筛中吸附行为的分子模拟研究
党 宇,丁 雪,刘熠斌,冯 翔
(中国石油大学(华东)重质油国家重点实验室,山东 青岛 266580)
通过分子模拟的方法研究了劣质重油中典型的氮化物(吲哚)在4种不同硅铝比的FAU分子筛上的吸附行为。结果表明:在温度823 K下,吲哚在4种FAU分子筛上的吸附量随着压力的增大而增加,并在800 kPa左右达到饱和状态;在一定范围内,随着硅铝比的增大,分子筛孔道的自由体积增加,吲哚在FAU分子筛上的饱和吸附量增加;通过Langmuir关联,证明了吲哚在FAU分子筛上的吸附属于单分子层吸附;根据分子模拟结果可从微观角度解释FAU分子筛硅铝比对吲哚吸附性能的影响。
吲哚 FAU分子筛 吸附 分子模拟
催化裂化是重油轻质化的重要手段。随着原油的日益重质化和劣质化,催化裂化逐步掺炼渣油、焦化蜡油等劣质原料[1]。这类劣质原料中含有大量的含氮化合物,而氮化物对催化裂化过程会产生不利影响,包括:① 使催化裂化催化剂中毒失活,主要通过诱导效应将催化剂酸中心中和,以及含氮的大分子稠环化合物形成焦炭并将酸中心覆盖[2-3];② 影响催化裂化产物分布,产物中总液体收率下降、汽油收率降低、干气和焦炭产率增加[4]。随着催化裂化劣质原料加工量的增加,氮化物对催化裂化过程的影响变得更加突出。
分子模拟是现阶段广泛应用的一种高效模拟方法,其基本思想是依据相关分子理论构建一个模型,建立一种通过软件模拟结果来代替实验测量的研究方法。它主要采用模拟的手段来研究与分子有关的各种信息,主要的优势在于可以降低实验成本,具有很高的安全性,实现通常条件下难以进行的实验,以及研究快速的反应和变化等。国内外很多学者采用模拟的方法研究了催化裂化原料中氮化物的吸附性质。沈喜洲等[5-6]采用GCMC方法研究胺类、喹啉类氮化物在FAU分子筛上的吸附行为,得到了吸附热与胺类碱性分子大小的关系。王丽新等[7]采用量子化学的方法研究了溶剂的极性和含氮化合物的碱性对氮化物与H+质子结合能的影响,结果表明溶剂极性越大,氮化物碱性越强,结合能越大。Injan等[8]采用DFT和ONIOM方法,考察了长程静电相互作用对吡啶在FAU分子筛上吸附热的影响,吸附热的模拟结果与实验值吻合较好。Kassab等[9]采用从头算的方法研究了吡啶和4,4′-二吡啶在氧化铝表面L酸位的吸附性质,频率分析结果表明,氧化铝表面的L酸位与碱性氮化物有较强相互作用,模拟结果与实验值十分接近。截至目前,关于吲哚在FAU分子筛上的吸附研究还未见报道。
本课题采用分子模拟的方法研究在催化裂化温度(823 K)下,劣质重油中典型的氮化物(吲哚)在不同硅铝比FAU分子筛上的吸附性质,获得吸附等温线、吸附位和吸附构型等信息,考察硅铝比对吸附性能的影响,从理论上预测氮化物在FAU分子筛上的吸附行为,为设计适宜硅铝比的FAU分子筛提供重要指导。
1 计算模型与方法
1.1 分子筛模型
FAU分子筛的主要结构是八面沸石笼,孔径为0.74 nm,可以让很多种有机分子进入孔道内。FAU型分子筛的框架类似于金刚石密堆六方晶系结构,用4个六方柱笼将5个β 笼联在一起,其中1个 β 笼居中心,其余4个 β笼位于正四面体顶点,最终组成的晶体结构就是八面体沸石型晶体结构。八面沸石笼就是这种由六方柱笼和β笼形成的大笼,相通的孔窗为12元环。X型、Y型是FAU分子筛的两种主要类型,其区别在于硅铝比不同,X型FAU分子筛的硅铝比一般为1~1.5,Y型FAU分子筛的硅铝比为1.5~3.0。这两种类型的分子筛都是当前石油加工中最广泛使用的催化剂。
Tokuhiro等[10-11]研究了不同FAU分子筛的结构信息,并给出晶胞中各原子的坐标参数等数据,其中Na+的位置也被确定,如表1所示。

表1 Na+的位置坐标
本研究使用Materials Studio软件构建FAU分子筛模型,空间群为Fd-3m,a=b=c=25.028 nm,α=β=γ=90°,并根据文献[12]调整分子筛的硅铝比。引入Na+平衡电荷并指定各原子电荷,NaX(硅铝比为1.18)分子筛的单胞组成为:Na88Al88Si104O384,每个晶胞需要引入88个Na+来平衡骨架电荷,将I位、I′位和II位全部占满,8个Na+占据III位,分子筛中各原子电荷指定如下:Si(+2.4),Al(+1.7),Oz(-1.2),Na(+0.7)[13-14]。硅铝比为2.56的NaY分子筛的单胞组成为:Na54Al54Si138O384,每个晶胞需要引入54个Na+,将I位和I′位占满,6个Na+占据II位,分子筛中各原子的电荷指定如下:Si(+2.4),Al(+1.4),Oz(-1.2),Na(+1.0)[13-14]。硅铝比为3.00的NaY分子筛的单胞组成为:Na48Al48Si144O384,每个晶胞需要引入48个Na+,将I位和I′位占满,原子电荷指定如下:Si(+2.4),Al(+1.4),Oz(-1.2),Na(+1.0)[13-14]。全硅型分子筛的原子电荷指定如下:Si(+2.4),Oz(-1.2)。4种FAU分子筛的结构见图1。
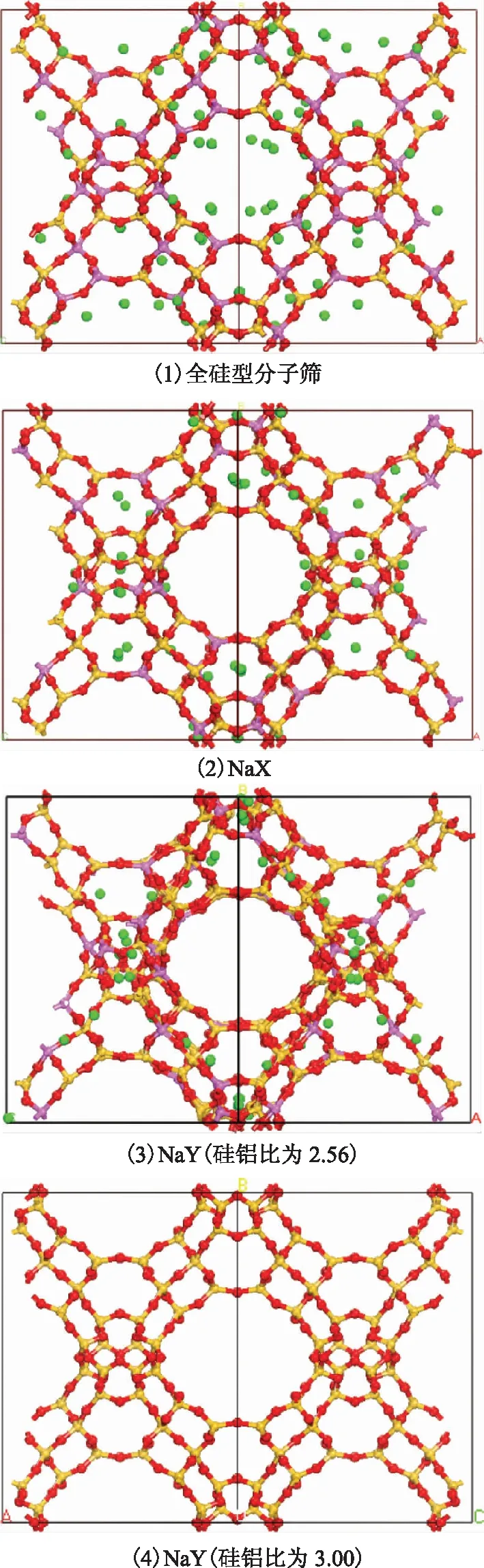
图1 4种FAU分子筛的结构模型●—O原子; ●—Si原子; ●—Al原子; ●—Na原子
1.2 模型验证
采用Materials Studio软件的Forcite模块对搭建的4种FAU分子筛进行结构优化,以获得各原子的合适位置和分子筛能量最低的结构。为了验证所搭建的分子筛模型的合理性,通过计算得到X射线衍射图谱(XRD),如图2所示。将其与国际沸石协会(IZA-SC)数据库中的相应分子筛的标准XRD图谱进行对比,发现两者具有相同的特征峰,表明搭建的4种FAU分子筛模型的结构合理。
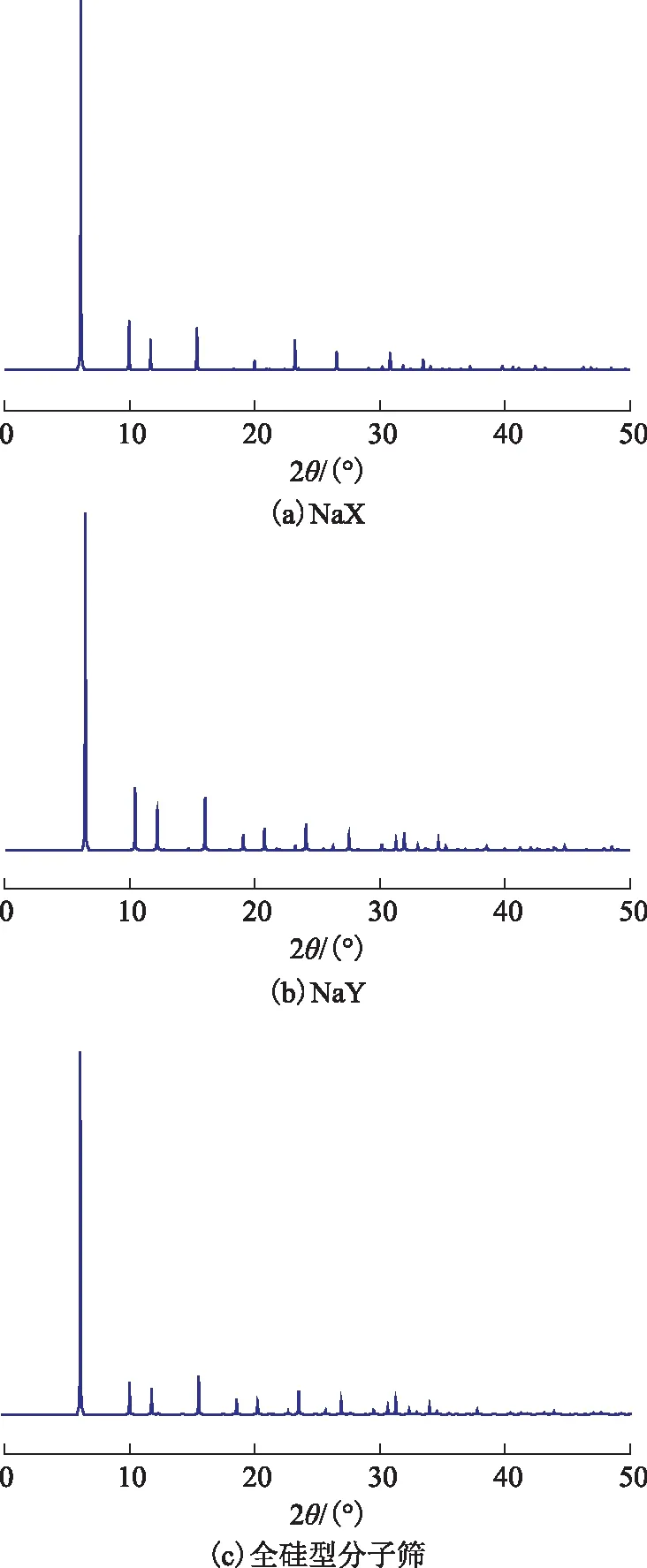
图2 不同FAU分子筛模型的XRD图谱
1.3 吲哚模型
吲哚是一种亚胺类化合物,具有弱碱性,其三维尺寸如下:a=0.810 5 nm,b=0.655 6 nm,c=0.228 5 nm。吲哚分子的最小截面尺寸为0.228 5 nm×0.655 6 nm,而FAU分子筛的孔道尺寸为0.74 nm×0.74 nm,因此吲哚分子理论上可以扩散进入FAU分子筛的孔道。使用Dmol3模块对吲哚分子进行结构优化,并计算各原子的ESP电荷,结果见表2。
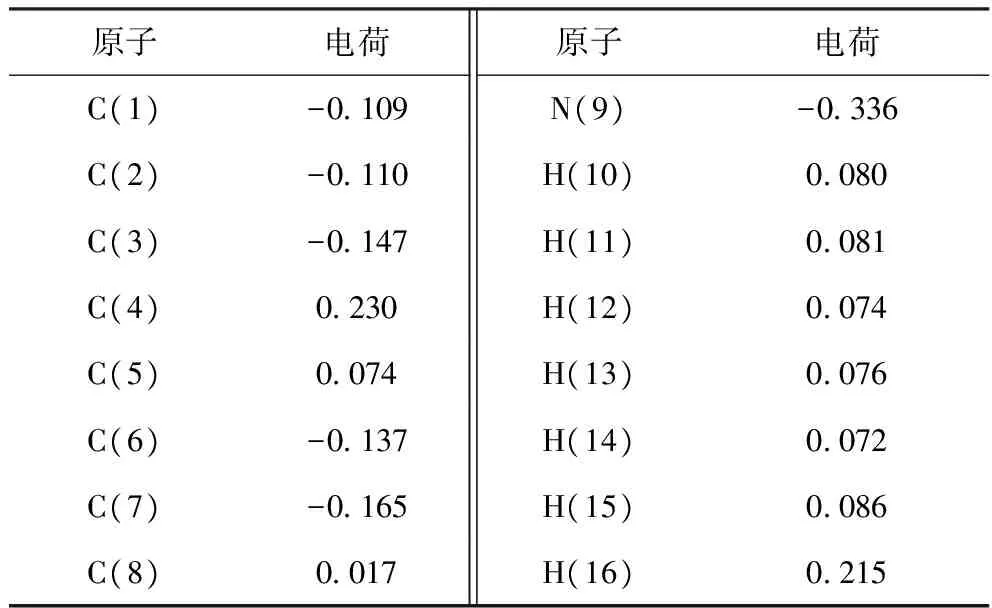
表2 吲哚中原子的ESP电荷
1.4 蒙特卡罗方法
蒙特卡罗(MC)方法是一种利用随机取样解决问题的计算机方法,其基本思想是建立一个能代表真实体系的概率模型或随机过程,通过对模型进行观察和抽样实验来得到所求问题的近似解,引入玻尔兹曼因子描述各种温度下的平均结构。简言之,就是用少数粒子长时间的平均行为来取代大量粒子的瞬间平均行为。系综是一定宏观条件下大量性质和结构完全相同的、处于各种运动状态的、各自独立的系统的集合,主要包括微正则系综(NVE)、正则系综(NVT)、巨正则系综(μVT)。当蒙特卡罗方法采用巨正则系综时,称为巨正则蒙塔卡罗(GCMC)方法,这意味着体系的粒子数不固定,但体系的化学势μ、体积V和温度T固定,吸附达到平衡时,骨架内吸附质的化学势和温度与骨架外的相等,进而可以研究在固定温度和压力下吸附质分子在分子筛内的吸附特性[15-16]。
2 模拟参数设置
Dmol3模块的相关参数设置如下:精度为Fine精度,选取局域密度近似泛函(LDA),收敛判据选择Fine精度,核处理选择考虑所有电子,数值基组选择DNP基组。Forcite模块中的算法选择Smart方法;计算静电相互作用(Electrostatic)和非键相互作用(van der Waals)的加和方法均选择原子截断(Atom based)算法。Sorption模块中的抽样方法选择Metropolis方法,精度选择Ultra-fine,分子筛骨架和吸附质分子之间包含两种相互作用,其数学表达式为:
(1)
式中:前半部分表示范德华力作用,后半部分表示静电相互作用;i和j表示不同原子;Rij表示原子间距;Dij和(R0)ij为Lennard-Jones参数,q表示原子所带电荷。
力场选择Compass力场,静电作用采用Ewald加和法处理,计算范德华作用的加和方法采用Atom based算法,非键截断值设置为1.25 nm,正好小于晶胞边长的一半(2.502 8 nm),平衡计算步数为106步,生产步数为107步。
3 结果与讨论
3.1 吸附等温线
首先计算在压力为0~1 MPa、温度为823 K的条件下,吲哚在全硅型分子筛、NaX分子筛和2种NaY分子筛中的吸附等温线,结果如图3所示。
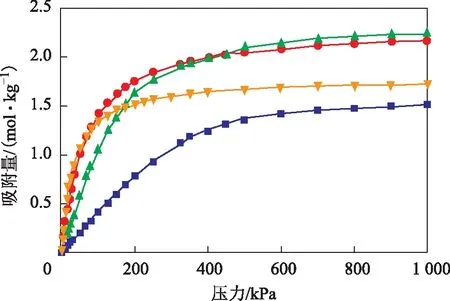
图3 吲哚在4种FAU分子筛中的吸附等温线▲—NaX; ●—NaY(硅铝比为2.56); 硅铝比为3.00); ■—全硅型分子筛
采用朗格缪尔(Langumir)吸附等温线模型[见式(2)] 对823 K下吲哚在4种分子筛上的吸附数据进行拟合,结果见表3。

(2)
式中:V为吸附量,mmol/g;Vm为饱和吸附量,mmol/g;B为吸附作用平衡常数,kPa-1;p为实验条件下平衡时的压力,kPa。
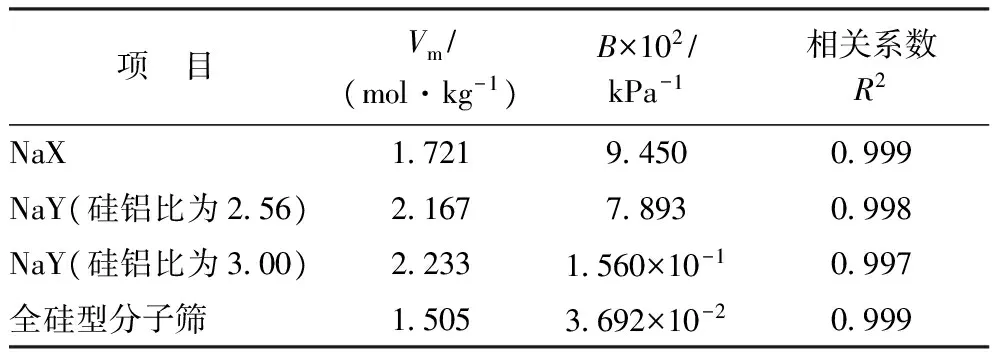
表3 823 K下吲哚在4种FAU分子筛中吸附的Langmuir拟合参数
由图3和表3可知:吲哚在4种FAU分子筛中的吸附等温线属于Ⅰ型等温线,是典型的单分子层吸附,该曲线可以用Langmuir方程很好地拟合,相关系数R2均大于0.99;在全硅型FAU分子筛中,吲哚的吸附量随着压力的增大缓慢增大,在800 kPa附近吸附达到饱和状态,饱和吸附量为1.505 mol/kg;而在NaX和两种NaY分子筛中,低压(0~200 kPa)时吲哚的吸附量随压力的增大而迅速增大,在压力达到800 kPa后,吲哚的吸附量基本不再随压力增大而增加,即分子筛吸附达到饱和状态,NaX分子筛、NaY分子筛(硅铝比为2.56)、NaY分子筛(硅铝比为3.00)的饱和吸附量分别为1.721,2.167,2.233 mol/kg。出现上述现象的原因主要为:在全硅型分子筛中,吲哚分子主要通过孔道的范德华力作用吸附,导致在低压范围内吸附量缓慢增加,饱和吸附量较低;对于NaX和NaY分子筛,除范德华力作用外,吲哚分子与Na+之间还存在较强的静电相互作用,使得在低压范围内吸附量随压力增大而迅速增加,并且饱和吸附量较高。
3.2 吸附位
通过分子筛和吸附质之间相互作用的能量分布曲线,可以得到吸附质分子在分子筛中的吸附位点和最可几分布位置[5]。图4为823 K、800 kPa下吲哚在4种FAU分子筛中吸附的能量分布曲线。由图4可知,4种FAU分子筛的能量分布曲线均只有一个吸附峰,NaX分子筛、NaY分子筛(硅铝比为2.56)、NaY分子筛(硅铝比为3.00)、全硅分子筛的出峰位置分别为-108,-96,-79,-58 kJ/mol 附近,表明吲哚在4种FAU分子筛的一个晶胞中只存在一种吸附位点,均在超笼的II位附近。

图4 吲哚在4种FAU分子筛中吸附的能量分布 —NaX; —NaY(硅铝比为2.56); 硅铝比为3.00); ●—全硅型分子筛
在823 K下,吲哚在4种FAU分子筛中吸附的密度场分布如图5所示(图中深蓝色区域表示吲哚分子在该位置的分布密度)。由图5可知,在823 K下,吲哚在4种FAU分子筛中吸附时,只分布在十二元环组成的超笼中,在六元环和四元环组成的孔道内没有分布。

图5 吲哚在4种FAU分子筛中吸附的密度场分布
3.3 吸附构型
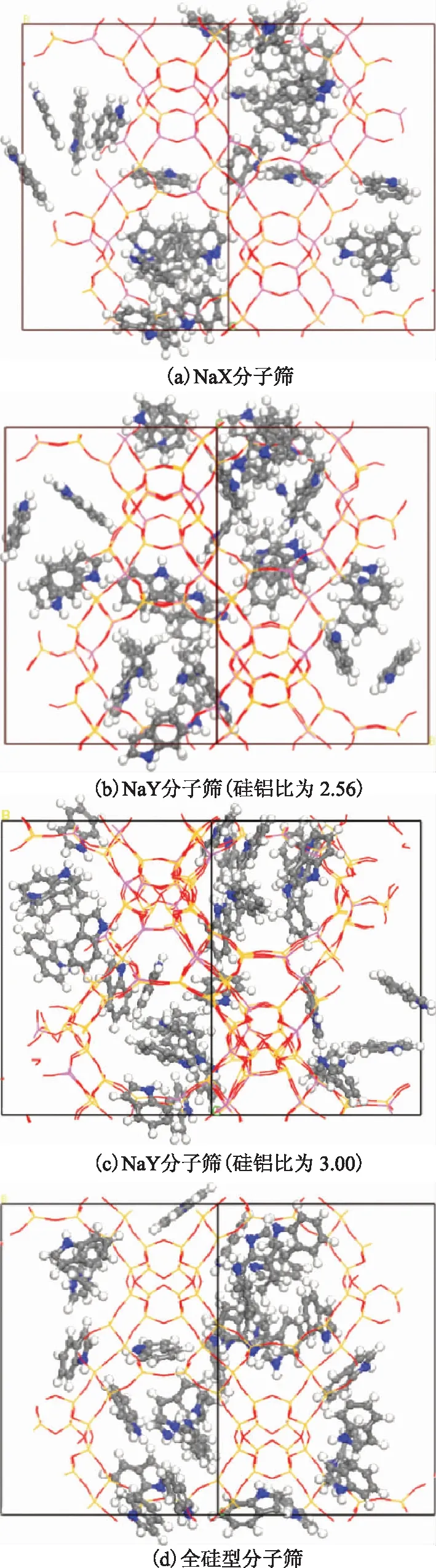
图6 吲哚在4种FAU分子筛中吸附的低能构象
为进一步分析吲哚分子在FAU分子筛中的吸附构型,研究了吲哚在4种FAU分子筛中吸附饱和时的最低能量构象,结果如图6所示(为了方便观察吲哚分子在分子筛中的分布,图中去掉了所有的Na+)。由图6可知,吲哚分子仅在分子筛的超笼中吸附,由于尺寸大小的限制使得吲哚分子在其它孔道中没有吸附,这一点和密度分布的结果是一致的。吲哚在NaX分子筛中的稳定吸附构型有两种,一种是以分子中的N原子所在的五元环朝向Na离子的形式吸附,这主要是由于N原子带0.336个单位的负电荷,与分子筛中的Na+存在较强的库仑作用;另一种构型是以吲哚分子中的苯环朝向Na+的形式吸附,主要是通过苯环上的π电子与分子筛中的Na+之间的静电相互作用形成π吸附[17]。吲哚在两种NaY分子筛上的吸附构型与NaX分子筛相似,同样只吸附在分子筛的超笼II位附近,并且也存在上述两种吸附构型。对于全硅分子筛,吲哚分子通过孔道的范德华作用吸附在超笼II位附近。
Fukui函数能够反映分子对电子得失的敏感性,Fukui函数值越大的位点,其对应的反应活性也越大[18-19]。吲哚分子的Fukui函数计算结果如图7所示,图中吲哚分子被一个3D等值面包围,以显示反应区域,等值面上不同的颜色表示电子密度不同,其中红色区域的电子密度高,反应活性高。由图7可知,吲哚分子上的高反应活性主要分布在两个区域,一个是在含N原子的五元环上,另一个是在苯环上,该结果验证了吲哚分子在NaX分子筛和NaY分子筛中的吸附构型。
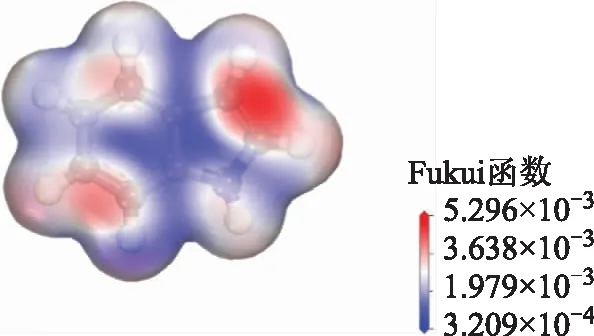
图7 吲哚分子的Fukui函数计算结果
3.4 硅铝比对吸附性能的影响
硅铝比的变化对于分子筛有两个方面的影响:一方面,分子筛中的非骨架阳离子会与极性分子之间发生较强的静电相互作用,由上文的能量分布分析结果可知,吲哚与NaX分子筛、NaY分子筛(硅铝比为2.56)、NaY分子筛(硅铝比为3.00)、全硅分子筛的相互作用能的出峰位置分别为-108,-96,-79,-58 kJ/mol 附近,表明吲哚分子与NaX和NaY分子筛的相互作用更强,吸附更稳定[20],而与全硅分子筛的相互作用较弱,这主要是由于在全硅分子筛中吲哚分子主要通过范德华作用吸附,而范德华作用与静电相互作用相比是一种弱相互作用;另一方面,随着阳离子数量的增加,分子筛孔道中更多的体积被阳离子占据,使得吸附质分子可以到达的位置被压缩,不利于吸附质分子在孔道中的吸附和扩散,导致吸附量降低。

图8 4种FAU分子筛晶胞中的自由体积
用Connolly表面方法[21]计算了4种FAU分子筛内吲哚可达到的体积(自由体积),结果见图8,其中绿色区域表示吲哚分子可以到达的体积,灰色区域表示分子筛骨架的体积。4种FAU分子筛的总体积均为15.677 6 nm3,NaX分子筛、NaY分子筛(硅铝比为2.56)、NaY分子筛(硅铝比为3.00)、全硅型分子筛的自由体积分别为6.209 4,8.081 3,8.921 3,9.293 9 nm3。
由于受Na+的影响,分子筛中吲哚分子可以到达的体积小于分子筛的实际孔体积,表4列出了4种FAU分子筛的几何数据[22]。从表4可以看出,随着硅铝比增大,分子筛骨架占有体积降低,自由体积增大,即吲哚分子可以到达的体积增大。这主要是由于NaX分子筛的硅铝比较低,填充的Na+数量较多,使得Na+占据了超笼中部分位点,不利于吲哚分子在孔道中的吸附;另外,NaX分子筛中的Na+部分填充在四元环附近(接近超笼的四元环 Ⅲ 位),而NaY分子筛中的Na+大多填充在六元环中心附近(主要在方钠石笼中,略微进入超笼的Ⅱ位)。NaX分子筛中的Na+主要分布在超笼孔口,导致孔口直径减小,不利于吲哚分子扩散进入孔道中,吸附阻力增大。而NaY分子筛由于硅铝比较大,Na+在孔口分布少,对吲哚分子的扩散影响较小,吲哚分子的吸附阻力小,因此吸附量较高。但是,硅铝比并不是越大越好,对于全硅型FAU分子筛,硅铝比为无穷大,分子筛中不需要补偿Na+,分子筛的自由体积最大,但是吲哚分子主要通过范德华作用吸附进入孔道,导致低压范围内吸附缓慢,同时饱和吸附量下降。

表4 4种FAU分子筛的几何数据
以上研究表明,硅铝比主要是通过影响Na+的数量和分布来影响吲哚的吸附性质。硅铝比较低时,Na+的数目较多,并且Na+在超笼孔口的分布不利于吲哚分子扩散进入笼中,吲哚分子的吸附阻力增大,吸附量下降。而硅铝比越高,填充的Na+数目越少,分子筛自由体积较大,并且Na+在超笼孔口分布均匀,吲哚分子的吸附和扩散的阻力小。但是并非硅铝比越大越好,因为硅铝比达到一定数值时,吲哚分子与分子筛的相互作用下降,吸附量下降。这说明不同的硅铝比对吲哚的吸附性质有较大的影响,在今后的研究工作中可以用此性质来设计适宜硅铝比的FAU分子筛。
4 结 论
(1)在823 K下,吲哚在4种FAU分子筛上吸附量随着压力的增大而增加。低压范围内,吲哚在全硅型分子筛中的吸附量随着压力的增大而缓慢增大,而在NaX和两种NaY分子筛中的吸附量随压力增加而迅速增大。吲哚在NaX分子筛、NaY分子筛(硅铝比为2.56)、NaY分子筛(硅铝比为3.00)、全硅分子筛中的饱和吸附量分别为1.721,2.167,2.233,1.505 mol/kg。
(2)通过对吲哚在4种FAU分子筛上的吸附等温线进行Langmuir关联后发现,4种分子筛的吸附等温线都属于Ⅰ型等温线,符合Ⅰ型吸附,是典型的单分子层吸附。
(3)硅铝比通过影响Na离子的数量和分布进而影响吲哚在分子筛的饱和吸附量,在一定范围内,随着硅铝比的增大,分子筛孔道的自由体积增加,吲哚分子的饱和吸附量增大。
[1] 袁起民,龙军,谢朝钢,等.高氮原料的催化裂化研究进展[J].化工进展,2008,27(12):1929-1936
[2] Corma A,Fornes V,Monton J B,et al.Catalytic cracking of alkanes on large pore,high SiO2Al2O3zeolites in the presence of basic nitrogen compounds:Influence of catalyst structure and composition in the activity and selectivity[J].Industrial & Engineering Chemistry Research,1987,26(5):882-886
[3] Caeiro G,Magnoux P,Lopes J M,et al.Kinetic modeling of the methylcyclohexane transformation over H-USY:Deactivating effect of coke and nitrogen basic compounds[J].Journal of Molecular Catalysis A:Chemical,2006,249(12):149-157
[4] 陈小博,孙金鹏,沈本贤,等.碱性氮化物对USY和ZSM-5型催化裂化催化剂催化性能的影响[J].中国石油大学学报:自然科学版,2012,36(5):164-168
[5] 沈喜洲,李梅青,周涵,等.NH3及胺类氮化物在八面沸石中吸附的分子模拟[J].计算机与应用化学,2011,28(1):117-120
[6] Shen Xizhou,Yan Fang,Li Meiqing,et al. Molecular simulation of adsorption of quinoline homologues on FAU zeolite[J].China Petroleum Processing and Petrochemical Technology,2016,18(4):110-116
[7] 王丽新,周涵,代振宇,等.石油中含氮化合物碱性的分子模拟研究[J].计算机与应用化学,2009,26(8):971-974
[8] Injan N,Pannorad N,Probst M,et al.Pyridine adsorbed on H-Faujasite zeolite:Electrostatic effect of the infinite crystal lattice calculated from a point charge representation[J].International Journal of Quantum Chemistry,2010,105(6):898-905
[9] Kassab E,Castellàventura M.Theoretical study of pyridine and 4,4′-bipyridine adsorption on the Lewis acid sites of alumina surfaces based on ab initio and density functional cluster calculations[J].Journal of Physical Chemistry B,2005,109(28):13716-13728
[10] Tokuhiro T,Iton L E,Peterson E M.Nuclear magnetic resonance study of Li-exchanged zeolites:Ⅰ.Cation spin dynamics:7Li in hydrated A,X,and Y zeolites[J].Journal of Chemical Physics,1983,78(12):7473-7485
[11] And M F,Lobo R F.Characterization of Li cations in zeolite LiX by solid-state NMR spectroscopy and neutron diffraction[J].Chemistry of Materials,1998,10(10):2197-2204
[12] 黄孟凯.稠环芳烃在Y分子筛中吸附和扩散的模拟研究[D].上海:华东理工大学,2013
[13] 黄艳芳,马正飞,刘晓勤,等.LTA和FAU型分子筛吸附CO2力场的研究[J].计算机与应用化学,2010,27(6):759-764
[14] Olson D H.The crystal structure of dehydrated NaX[J].Zeolites,1995,15(5):439-443
[15] Hou Tingjun,Zhu Lili,Li Youyong,et al.The localization and adsorption of benzene and propylene in ITQ-1 zeolite:Grand canonical Monte Carlo simulations[J].Journal of Molecular Structure:THEOCHEM,2001,535(1):9-23
[16] Smit B.Simulating the adsorption isotherms of methane,ethane,and propane in the zeolite silicalite[J].Journal of Physical Chemistry,1995,99(15):5597-5603
[17] Jobic H,Fitch A N.Vibrational study of benzene adsorbed in NaY zeolite by neutron spectroscopy[J].Studies in Surface Science & Catalysis,1997,105:559-566
[18] Langenaeker W,Demel K,Geerlings P.Quantum-chemical study of the Fukui function as a reactivity index:Part 2.Electrophilic substitution on mono-substituted benzenes[J].Journal of Molecular Structure:THEOCHEM,1991,234(91):329-342
[19] Langenaeker W,Demel K,Geerlings P.Quantum-chemical study of the Fukui function as a reactivity index:Part 3.Nucleophilic addition toα,β-unsaturated compounds 1[J].Journal of Molecular Structure:THEOCHEM,1992,259(92):317-330
[20] 孙晓岩,张达,项曙光,等.苯与乙烯在ZSM-5分子筛内吸附行为的Monte Carlo研究[J].计算机与应用化学,2013,30(5):454-458
[21] Connolly M L.Computation of molecular volume[J].Journal of the American Chemical Society,1985,107(5):1118-1124
[22] 丁静,胡玉坤,杨晓西,等.水在ZSM-5型分子筛中吸附的Monte Carlo模拟[J].化工学报,2008,59(9):2276-2282
MOLECULARSIMULATIONOFADSORPTIONBEHAVIOROFINDOLEINFAUZEOLITESWTBZ
Dang Yu, Ding Xue, Liu Yibin, Feng Xiang
(StateKeyLaboratoryofHeavyOilProcessing,ChinaUniversityofPetroleum(EastChina),Qingdao,Shandong266580)
Molecular simulation is used to study the adsorption of indole in four different Si/Al ratio FAU zeolites.The results show that all the adsorption capacities of indole in four FAU zeolites at the 823 K increase with increasing adsorption pressure and come to a plateau at about 800 kPa.Within a certain experimental range,the greater the Si/Al ratio,the greater the indole adsorption amount in FAU zeolites due to enlarged free space.Langmuir correlations prove that the adsorption of indole in the four FAU zeolites is a monolayer adsorption.The simulated results show that the effect of Si/Al ratio on the adsorption performance of indole in FAU zeolites can be explained from the microscopic point of view.
indole; FAU zeolite; adsorption; molecular simulation
2017-05-31;修改稿收到日期2017-07-06。
党宇,硕士研究生,主要从事石油加工和分子模拟方面的研究工作。
刘熠斌,E-mail:liuyibin@upc.edu.cn。
山东省自然科学基金项目(ZR2014BL015,ZR2016BB16);国家自然科学基金项目(21606254)。
