维生素B12依赖型甲基丙二酸血症一家系临床表型和基因突变分析及疗效评价
2017-11-21利婧孙毅明欧俐羽朱瑜龄王倞李欢张成
利婧 孙毅明 欧俐羽 朱瑜龄 王倞 李欢 张成
维生素B12依赖型甲基丙二酸血症一家系临床表型和基因突变分析及疗效评价
利婧 孙毅明 欧俐羽 朱瑜龄 王倞 李欢 张成
目的探讨维生素B12依赖型甲基丙二酸血症家系临床特点、基因突变和维生素B12治疗效果.方法采集一维生素B12依赖型甲基丙二酸血症家系共4名成员临床资料,抽取外周静脉血行血浆氨基酸和酰基肉碱谱分析及基因检测,评价维生素B12治疗效果.结果家系中先证者12岁发病,以学习成绩下降、性格改变为首发症状,病程中出现幻觉、双下肢无力;先证者之弟主要表现为易怒、学习成绩较差.经血浆氨基酸和酰基肉碱谱分析,先证者及其弟血浆丙酰肉碱、丙酰肉碱/乙酰肉碱比值升高,尿液甲基丙二酸水平升高.基因检测显示,先证者及其弟均存在MMACHC基因复合杂合突变c.482G>A(p.Arg161Gln)和c.609G>A(p.Trp203X),其父携带MMACHC基因错义突变c.482G>A(p.Arg161Gln),其母携带MMACHC基因无义突变c.609G>A(p.Trp203X).经维生素B12治疗后均症状好转.结论晚发型维生素B12依赖型甲基丙二酸血症系MMACHC基因复合杂合突变所致,维生素B12治疗反应良好,早期诊断、及时治疗对患者预后意义重大.
甲基丙二酸; 代谢缺陷,先天性; 维生素B12; 表型; 基因; 突变; 系谱
甲基丙二酸血症(MMA)cblC型系MMACHC基因突变导致其编码的cblC蛋白缺乏,使L⁃甲基丙二酸无法转变为琥珀酸而在血液中蓄积,是临床最常见的维生素B12代谢障碍性疾病,呈常染色体隐性遗传,发病率为1/10万~1/6万[1].根据发病年龄,可以分为早发型(1岁内发病)和晚发型甲基丙二酸血症.迄今文献报道仅约10%患者为晚发型甲基丙二酸血症[2⁃3].维生素B12依赖型甲基丙二酸血症临床罕见,通常于儿童期、青少年期或成年期发病,主要表现为精神症状、意识障碍和神经系统症状等,经及时治疗预后较好[4⁃5],属可治性神经系统遗传性疾病.本研究报道一晚发型甲基丙二酸血症家系,总结其临床表现、血液化学、基因检测和维生素B12治疗后转归,探讨维生素B12治疗MMAcblC型的作用机制,以期提高儿科和神经科医师对甲基丙二酸血症的认识.
对象与方法
一、研究对象
本研究MMAcblC型患者于中山大学附属第一医院神经科就诊,其家系2代共4名成员(图1),汉族,其中2例符合常染色体隐性遗传特点.
先证者 女性,13岁,主因性格改变1年、双下肢无力2个月,于2016年11月24日至我院神经科门诊就诊.患者1年前无明显诱因出现易激惹、易哭闹,学习成绩下降,未予重视;2个月前出现双下肢无力,勉强能够独立行走,上楼梯费力,无肢体麻木,双上肢未见异常;1月余前出现幻觉,以幻视为主,逐渐进展至行走不能.1个月前曾至外院就诊,诊断为"可疑线粒体脑肌病或脑炎",予静脉注射免疫球蛋白(IVIg)和甲泼尼龙等治疗(具体剂量不详),症状无明显好转,为求进一步诊断与治疗,遂至我院就诊.患儿足月顺产,单胎,生长发育和智力发育正常;父母身体健康,无相似临床症状,否认近亲婚配.门诊体格检查:身高和体重于同龄儿童正常范围.神经系统检查:高级智能和脑神经检查未见异常,双侧小腿肌萎缩,双下肢近端肌力3级、远端4级,肌张力降低,双上肢肌力5级,肌张力正常;双下肢痛触觉和关节位置觉减退,四肢腱反射未引出,双下肢Babinski征阳性,脑膜刺激征阴性.实验室检查:血尿便常规、肝肾功能试验、血液化学、免疫学指标、血氨、血清维生素B12均于正常值范围,血清同型半胱氨酸(Hcy)165.30 μmol/L(5~20 μmol/L).影像学检查:胸部X线未见明显异常.头部MRI显示轻度脑萎缩,尤以白质显著,双侧侧脑室旁长T1、长T2信号,扩散加权成像(DWI)呈颞顶叶放射冠低信号,表观扩散系数(ADC)升高(图2).磁共振波谱(MRS)显示,N⁃乙酰天冬氨酸(NAA)峰、肌酸(Cr)峰、胆碱(Cho)峰峰值降低(图3).电生理学检查:心电图未见明显异常.脑电图显示中度异常,以θ波为主要背景波.
患儿Ⅱ2 男性,10岁,先证者之弟,于2016年12月6日至我院神经科门诊就诊.患儿足月顺产,单胎,18个月方可独立行走和会说话,智力发育较同龄正常儿童差,目前读小学,学习成绩较差,易怒,未予重视.门诊体格检查:身高和体重于同龄儿童正常范围.神经系统检查:注意力和计算力较差,语言复述能力较差,脑神经检查未见明显异常;四肢肌力5级、肌张力正常,感觉系统未见异常,腱反射降低,病理反射未引出,脑膜刺激征阴性.实验室检查:血尿便常规、肝肾功能试验均于正常值范围,血氨 51 μmol/L(10~47 μmol/L),血清维生素B12于正常值范围,血清同型半胱氨酸84.20 μmol/L.
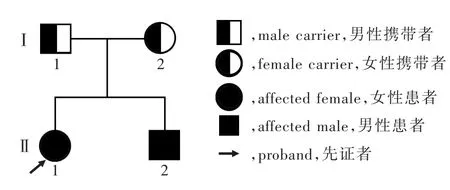
图1 MMAcblC型家系图Figure 1 The pedigree of MMAcblC.
二、研究方法
1.血浆氨基酸和酰基肉碱谱分析 采集先证者及其弟外周静脉血各2 ml制备干血滤纸,API3200液质联用仪(美国SCIEX公司)采用串联质谱法行血浆氨基酸和酰基肉碱谱分析.采集先证者及其弟尿液制备3张尿液滤纸片,7890/5975C气相质谱联用仪(美国Agilent公司)行尿液气相色谱质谱分析,检测尿液各种有机酸之间的比值.
2.目标区域捕获测序与数据分析 经先证者父母同意,采集该家系4名成员外周静脉血各2 ml,行甲基丙二酸血症相关二代基因测序(NGS).(1)基因组DNA提取和基因组文库制备:2 ml外周静脉血置于乙二胺四乙酸(EDTA)抗凝管中,采用QIAmp DNA Mini Kit试剂盒(德国QIAGEN公司)提取基因组 DNA. 取 3 μ g DNA,以 Tris⁃乙 二 胺 四 乙 酸(EDTA)缓冲液稀释至30 ng/μl,采用Covris⁃S220超声仪(美国Covaris公司)将样本DNA片段化为长度150 bp,采用标准DNA文库构建试剂盒(北京迈基诺基因科技股份有限公司)进行片段末端修复加"腺嘌呤"和产物纯化,经聚合酶链反应(PCR)扩增后,采用Nanodrop 2000紫外分光光度计(美国Therom公司)进行DNA文库定量.(2)目标区域捕获测序:采用GenCap液相捕获目标基因技术对已知的176种代谢性疾病相关基因进行探针捕获,覆盖每种基因的外显子及其上下游内含子长度50 bp区域,生物素标记的探针与文库DNA杂交,链霉素标记的磁珠共价结合生物素标记的探针,抓取目的基因,磁力架吸附携带目的基因的磁珠,洗脱纯化,富集目的基因,再采用NextSeq 500高通量测序仪(美国Illumina公司)进行双端测序.(3)数据处理与分析:原始数据剔除接头和低质量短序列(<40 bp),BWA软件(http://bio-bwa.sourceforge.net/)将预处理的数据与hg19人类基因组数据库(http://hgdownload.cse.ucsc.edu/)进行比对,GATK3.3.0软件包(https://www.broadinstitute.org/gatk)校准并重新匹配至参考序列,单碱基突变采用GATK UnifiedGenotyper分析,单核苷酸多态性(SNP)和插入/缺失突变采用共识编码序列数据库(CCDS)、hg19人基因组数据库、单核苷酸多态性数据库信息进行注释,确定候选变异位点,SIFT预测软件(http://sift.jcvi.org)对变异位点进行生物信息学分析.
3.Sanger测序验证 据基因变异位点设计上下游引物,由赛默飞世尔科技(中国)有限公司合成,上游引物序列:5'⁃CCTCCATGACCTTGCTTTTC⁃3',下游引物序列:5'⁃GGGATGCAGGTGGTGAGAC⁃3'.PCR反应体系共25 μl,反应条件为95℃ 10 min;94℃ 30 s、64℃ 30 s、72℃ 45 s,共3个循环;94℃ 30 s、62℃ 30 s、72℃ 45 s,共5个循环;94 ℃ 30 s、60℃ 30s、72℃ 45s,共 10 个循环;94℃30 s、58 ℃ 30 s、72 ℃ 45 s,共 17 个循环;最终72℃ 5 min;4℃冷却备用.然后采用ABI3130xl测序仪[赛默飞世尔科技(中国)有限公司]将PCR扩增产物进行毛细管电泳,测序结果与标准参考序列(NM_001330540.1)进行比对.
结 果
一、临床特征
该家系共2代4名成员,男性2名,女性2名;先证者12岁发病,主要表现为性格改变、学习成绩下降,病程中出现幻觉、双下肢无力症状,病程1年;先证者之弟自幼表现为易怒、智力低下,未明显累及运动系统.
二、血浆氨基酸和酰基肉碱谱分析
先证者血浆甲硫氨酸为7.80 μmol/L(8.50~50.00μmol/L),丙二酸0.60μmol/L(0~0.30μmol/L),丙酰肉碱(C3)6.39 μmol/L(0.30~5.00 μmol/L),丙酰肉碱/乙酰肉碱(C2)比值为0.41(0.02~0.25);尿液甲基丙二酸水平为 63.70 μmol/L(0~4 μmol/L).先证者之弟血浆丙酰肉碱5.29 μmol/L,C3/C2比值为 0.59,天冬氨酸为 62.60 μmol/L(18~47 μmol/L),精 氨 酸 95.50 μ mol/L(11~69 μ mol/L),丙 氨 酸576.70 μmol/L(210~545 μmol/L),谷氨酰胺水平为702 μmol/L(249~579 μmol/L);尿液甲基丙二酸为75 μmol/L.


图2 头部MRI检查所见 2a 横断面T2WI显示,轻度脑萎缩,尤以白质显著(箭头所示),双侧侧脑室轻度扩大 2b 冠状位FLAIR成像显示,双侧侧脑室旁异常高信号(箭头所示)2c 横断面DWI显示,双侧颞枕叶深部侧脑室旁异常低信号(箭头所示)2d 横断面ADC图显示病变呈高信号(箭头所示) 图3 头部MRS显示,左侧侧脑室旁NAA峰、Cr峰、Cho峰和NAA/(Cho+Cr)比值降低,考虑白质髓鞘化不良,提示代谢性疾病Figure2 Head MRI findings AxialT2WI showed mild encephalatrophy,espically in the white matter(arrows indicate),and slight enlargment of the bilateral ventricles (Panel 2a). Coronal FLAIR image showed abnormal hyperintense signals in bilateral periventricular area(arrows indicate,Panel 2b).Axial DWI showed abnormal hypointense signal in the deep part of bilateral temporo⁃occipital lobe beside lateral ventricle(arrows indicate,Panel 2c).Axial ADC showed hyperintensities in the lesion(arrow indicates,Panel 2d). Figure 3 MRSshoweddecreaseofNAA,Cr,ChoandNAA/(Cho+Cr)inleft periventricular regions,which reflected of neuronal damage and malformation in white matter myelination and suggested metabolic disease.
三、基因检测
先证者存在MMACHC基因复合杂合突变,包括错义突变c.482G>A(p.Arg161Gln)和无义突变c.609G>A(p.Trp203X),符合MMAcblC型的诊断(图4a).先证者之弟亦存在MMACHC基因复合杂合突变,包括错义突变c.482G>A(p.Arg161Gln)和无义突变c.609G>A(p.Trp203X,图4b).进一步对其父母行MMACHC基因检测,提示错义突变c.482G>A(p.Arg161Gln)源自其父(图4c)、无义突变c.609G>A(p.Trp203X)源自其母(图4d).2例患者最终明确诊断为MMAcblC型合并高同型半胱氨酸血症.
四、治疗
先证者明确诊断后即予甲钴胺1 mg/d肌肉注射,左卡尼汀2 g/d静脉滴注,以及利培酮0.50 g/次、2次/d口服控制幻视,共住院13 d,出院时幻视消失,双下肢肌力4级、肌张力降低,搀扶可行走.出院后予以甲钴胺1 mg/次、3次/d和左卡尼汀1 g/次、2次/d口服,并嘱低蛋白饮食.出院后3个月门诊随访,临床症状好转,双下肢肌力5-级,可独立行走,未再出现幻觉,复查血浆氨基酸和酰基肉碱谱分析,丙酰肉碱降至正常值范围,C3/C2比值0.27.先证者之弟明确诊断后予甲钴胺1 mg/次、3次/d和左卡尼汀1 g/次、2次/d口服,嘱低蛋白饮食.3个月后门诊随访,语言复述能力较前好转,注意力和计算力欠佳.
讨 论
甲基丙二酸血症是维生素B12代谢障碍性疾病,可以根据维生素B12在细胞内代谢障碍累及环节分型(图5[4],表1).人体从食物中摄取的游离维生素B12在体内与内因子结合形成稳定复合物,在回肠远端与肠黏膜细胞膜受体结合(维生素B12转运蛋白复合体)而被吸收.维生素B12进入细胞后自其转运蛋白复合体释放至细胞质内,在cblC蛋白催化下形成Co(Ⅱ)cbl蛋白(即带2价电子的氧化态cbl蛋白),一部分转运至线粒体内转化为腺苷钴胺素(AdoCbl),参与线粒体内支链氨基酸和奇数链脂肪酸代谢,人体必需的支链氨基酸和奇数链脂肪酸在分解代谢过程中生成甲基丙二酸,在甲基丙二酰辅酶A变位酶(MUT)和AdoCbl共同催化下生成琥珀酰辅酶A,参与三羧酸循环,若MUT或AdoCbl代谢异常,可以导致甲基丙二酸、丙酸等中间代谢产物堆积,引起单纯型甲基丙二酸血症,包括cblA型、cblB型和mut型3种亚型;另一部分在细胞质内转化为甲钴胺(MeCbl),参与催化同型半胱氨酸转化为甲硫氨酸[4],若同时出现AdoCbl和MeCbl合成不足,可以影响同型半胱氨酸和甲基丙二酸的代谢,使甲基丙二酸和同型半胱氨酸蓄积[1],导致甲基丙二酸血症合并高同型半胱氨酸血症,包括cblC型、cblD型、cblF型和cblJ型[6⁃7],尤以cblC型常见[2,8].
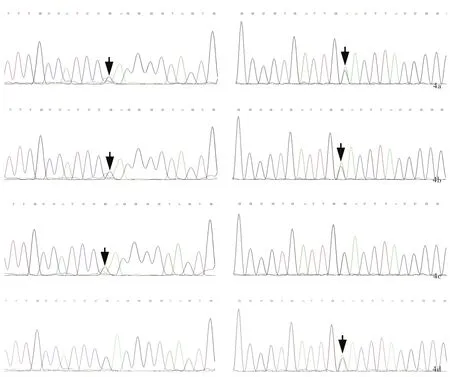
图4 MMACHC基因检测所见 4a 先证者存在MMACHC基因复合杂合突变,包括错义突变c.482G>A(p.Arg161Gln,箭头所示,左图)以及无义突变c.609G>A(p.Trp203X,箭头所示,右图)4b 先证者之弟存在MMACHC基因复合杂合突变,包括错义突变c.482G>A(p.Arg161Gln,箭头所示,左图)以及无义突变c.609G>A(p.Trp203X,箭头所示,右图) 4c 先证者之父亲存在MMACHC基因错义突变c.482G>A(p.Arg161Gln,箭头所示) 4d 先证者之母存在MMACHC基因无义突变c.609G>A(p.Trp203X,箭头所示)Figure 4 Analysis of MMACHC gene test Proband had compound heterozygous mutation in MMACHC gene,including missense mutation c.482G>A(p.Arg161Gln,arrow indicates,left panel)and nonsense mutation c.609G>A(p.Trp203X,arrow indicates,right panel,Panel 4a).Proband's younger brother had the same compound herterozygous mutation as the proband(arrows indicate,Panel 4b).The proband's father canrried MMACHC gene missense mutation c.482G>A(p.Arg161Gln,arrow indicates,Panel 4c).The proband's mother carried MMACHC gene nonsense mutation c.609G>A(p.Trp203X,arrow indicates,Panel 4d).
大多数甲基丙二酸血症为早发型,1岁内发病,表现为喂养困难、生长发育迟滞、癫发作、肌张力降低、酮症酸中毒等,除神经系统症状外,还有肝肾功能障碍表现等,若无早期干预,病死率较高[9⁃10],因此,国外和国内部分地区已将甲基丙二酸血症作为新生儿出生缺陷的常规筛查项目[8,11].晚发型甲基丙二酸血症患者少见,临床表现多样,既往有文献报道,1岁至学龄期前发病的患儿以溶血性尿毒综合征(HUS)和肺动脉高压为主要表现,而青少年期或成年期发病的患者则以不典型神经精神症状为主要表现或仅表现为学习困难、情绪障碍等,还可合并共济失调、厌食、癫发作等,临床明确诊断困难,常延误治疗[12⁃13].本研究家系中先证者以学习成绩下降、性格改变为首发症状,病程中出现幻觉,逐渐出现双下肢无力,体格检查可见双下肢肌力下降、下肢深浅感觉对称性减弱、腱反射减退或消失等周围神经损害体征,均支持晚发型甲基丙二酸血症的诊断,进一步行遗传代谢性疾病的血浆氨基酸和酰基肉碱谱分析,明确诊断.与既往文献报道的晚发型甲基丙二酸血症相似,先证者之弟仅出现学习成绩差、易怒等症状,一直未引起重视,待先证者明确诊断后,行基因检测才确定亦存在MMACHC基因突变.先证者头部MRI突出表现为脑萎缩、白质髓鞘化不良,与既往文献报道的甲基丙二酸血症典型影像学改变相符[13⁃16].

图5 维生素B12在细胞内代谢障碍累及环节以及甲基丙二酸血症各亚型[4]:cblA型、cblB型和mut型仅累及AdoCbl或MUT蛋白,引起单纯甲基丙二酸血症;cblC型、cblD型和cblF型导致AdoCbl和MeCbl蛋白合成障碍,引起甲基丙二酸血症合并高同型半胱氨酸血症Figure 5 Links and different subtypes of MMA caused by intracellular metabolism disorder of vitamin B12[4].cblA type,cblB type and mut type only affect AdoCbl and MUT protein,and cause simple MMA.cblC type,cblD type and cblF type induce the disorder of anabolism of AdoCbl and MeCbl protein and result in MMA complicated with hyperhomocysteinemia.

表1 甲基丙二酸各亚型相关基因Table 1. Different type of MMA and corresponding gene
MMAcblC型致病基因为MMACHC基因,定位于1p34.1,共包含5个外显子,全长10 800 bp,编码282个氨基酸残端蛋白即cblC蛋白,该蛋白在催化维生素B12向AdoCbl和MeCbl转化过程中发挥重要作用[2].本研究家系中先证者及其弟经基因检测明确诊断为MMAcblC型,系MMACHC基因复合杂合突变所致.其中,c.609G>A为无义突变,可使第203位色氨酸转录提前终止,该变异位点为东亚地区最常见变异位点,占40%~60%[8,17⁃19],该位点纯合突变多与早发型甲基丙二酸血症有关[19];c.482G>A为错义突变,可使第161位氨基酸由精氨酸突变为谷氨酰胺.Liu等[17]报告8例携带c.482G>A突变的患者,仅1例于4岁前发病,余均于4岁后发病,提示携带错义突变c.482G>A的患者临床表型多为晚发型.
本研究家系中先证者一经明确诊断即予甲钴胺、左卡尼汀治疗,早期即有效控制症状,治疗3个月后复查血浆丙酰肉碱和C3/C2比值明显下降,提示为MMAcblC型.既往研究显示,多数晚发型MMAcblC型为维生素B12依赖型,建议早期肌肉注射或皮下注射维生素B12(1 mg/d),同时予低蛋白饮食(每日蛋白质摄入量<0.80 g/kg),补充左旋肉碱[50~100 mg/(kg.d)],提高蛋氨酸水平、降低同型半胱氨酸和甲基丙二酸水平,改善临床症状[4,20⁃21].
晚发型甲基丙二酸血症临床罕见,但属可治性遗传性疾病,对维生素B12治疗反应良好,如果能够及时识别其不典型临床表现,通过特异性生物学指标和基因检测早期诊断、及时治疗,对患者预后意义重大.
[1]Carrillo⁃Carrasco N,Venditti CP.Combined methylmalonic acidemiaand homocystinuria,cblC type.Ⅱ:complications,pathophysiology,and outcomes.J Inherit Metab Dis,2012,35:103⁃114.
[2]Lerner⁃Ellis JP,Tirone JC,Pawelek PD,Doré C,Atkinson JL,Watkins D,Morel CF,Fujiwara TM,Moras E,Hosack AR,Dunbar GV,Antonicka H,Forgetta V,Dobson CM,Leclerc D,Gravel RA,Shoubridge EA,Coulton JW,Lepage P,Rommens JM,Morgan K,RosenblattDS.Identification ofthe gene responsible for methylmalonic aciduria and homocystinuria,cblC type.Nat Genet,2006,38:957.
[3]Lerner⁃Ellis JP,Anastasio N,Liu J,Coelho D,Suormala T,Stucki M,Loewy AD,Gurd S,Grundberg E,Morel CF,Watkins D,Baumgartner MR,Pastinen T,Rosenblatt DS,Fowler B.Spectrum of mutations in MMACHC,allelic expression,and evidence forgenotype⁃phenotype correlations.Hum Mutat,2009,30:1072⁃1081.
[4]Carrillo⁃CarrascoN,ChandlerRJ,Venditti CP.Combined methylmalonic acidemia and homocystinuria,cblC type.Ⅰ:clinicalpresentations,diagnosisand management.JInherit Metab Dis,2012,35:91⁃102.
[5]Ben⁃Omran TI,Wong H,Blaser S,Feigenbaum A.Late⁃onset cobalamin⁃C disorder:a challenging diagnosis.Am J Med Genet A,2007,143A:979⁃984.
[6]Coelho D,Kim JC,Miousse IR,Fung S,du Moulin M,Buers I,Suormala T,Burda P,Frapolli M,Stucki M,Nürnberg P,Thiele H,Robenek H,Höhne W,Longo N,Pasquali M,Mengel E,Watkins D,Shoubridge EA,Majewski J,Rosenblatt DS,Fowler B,Rutsch F,Baumgartner MR.Mutations in ABCD4 cause a new inborn error of vitamin B12metabolism.Nat Genet,2012,44:1152⁃1155.
[7]Yu HC,Sloan JL,Scharer G,Brebner A,Quintana AM,Achilly NP,ManoliI,Coughlin CR 2nd,GeigerEA,Schneck U,Watkins D,Suormala T,Van Hove JL,Fowler B,Baumgartner MR,Rosenblatt DS,Venditti CP,Shaikh TH.An X⁃linked cobalamin disorder caused by mutations in transcriptional coregulator HCFC1.Am J Hum Genet,2013,93:506⁃514.
[8]Han B,Cao Z,Tian L,Zou H,Yang L,Zhu W,Liu Y.Clinical presentation,gene analysis and outcomes in young patients with early⁃treated combined methylmalonic acidemia and homocysteinemia (cblC type)in Shandong province,China.Brain Dev,2016,38:491⁃497.
[9]Martinelli D,Deodato F,Dionisi⁃Vici C.Cobalamin C defect:natural history,pathophysiology,and treatment.J Inherit Metab Dis,2011,34:127⁃135.
[10]Huemer M,Kožich V,Rinaldo P,Baumgartner MR,Merinero B,Pasquini E,Ribes A,Blom HJ.Newborn screening for homocystinurias and methylation disorders:systematic review and proposed guidelines.J Inherit Metab Dis,2015,38:1007⁃1019.
[11]Weisfeld⁃Adams JD,Morrissey MA,Kirmse BM,Salveson BR,Wasserstein MP,McGuire PJ,Sunny S,Cohen⁃Pfeffer JL,Yu C,Caggana M,Diaz GA.Newborn screening and early biochemical follow ⁃up in combined methylmalonic aciduria and homocystinuria,cblC type,and utility ofmethionine as a secondary screening analyte.Mol Genet Metab,2010,99:116⁃123.
[12]Rahmandar MH,Bawcom A,Romano ME,Hamid R.Cobalamin C deficiency in an adolescent with altered mental status and anorexia.Pediatrics,2014,134:1709⁃1714.
[13]Huemer M,Scholl⁃Bürgi S,Hadaya K,Kern I,Beer R,Seppi K,Fowler B,Baumgartner MR,Karall D.Three new cases of late⁃onset cblC defect and review of the literature illustrating when to consider inborn errors of metabolism beyond infancy.Orphanet J Rare Dis,2014,9:161.
[14]Radmanesh A,Zaman T,Ghanaati H,Molaei S,Robertson RL,Zamani AA.Methylmalonic acidemia:brain imaging findings in 52 children and a review of the literature.Pediatr Radiol,2008,38:1054⁃1061.
[15]Liu Y,Yin J,Peng Y,Duan XM,Liu ZM,Yin GH.Brain imaging characters in 56 children with methylmalonic academia.Yi Xue Ying Xiang Xue Za Zhi,2013,23:165⁃169.[刘玥,阴捷,彭芸,段晓岷,刘志敏,尹光恒.56例儿童甲基丙二酸血症的脑改变影像学特点.医学影像学杂志,2013,23:165⁃169.]
[16]Lei RY,Liu YR,Ji YF,Ma XR,Wang JT,Cui HW,Wang Z,Wang CE,Zhang BA.Clinical features and gene analysis of three cases of late⁃onset methylmalonic aciduria,cblC type.Zhonghua Shen Jing Ke Za Zhi,2014,47:101⁃106[.雷如意,刘艳茹,籍炀飞,马兴荣,王景涛,崔红卫,王震,王长娥,张博爱.晚发型甲基丙二酸尿症cblC型三例临床特点和基因分析.中华神经科杂志,2014,47:101⁃106.]
[17]Liu MY,Yang YL,Chang YC,Chiang SH,Lin SP,Han LS,Qi Y,Hsiao KJ,Liu TT.Mutation spectrum of MMACHC in Chinese patients with combined methylmalonic aciduria and homocystinuria.J Hum Genet,2010,55:621⁃626.
[18]Cai AJ,Zong YN,Liu N,Wei ZL,Bai Y,Zhao ZH,Kong XD.MMACHC gene mutation analysis in the prenatal diagnosis of methylmalonic aciduria with homocystinuria.Zhonghua Jian Yan Yi Xue Za Zhi,2016,39:613⁃617[.蔡奥捷,宗亚楠,刘宁,魏振玲,白莹,赵振华,孔祥东.甲基丙二酸血症结合同型半胱氨酸血症家系MMACHC基因突变分析及在产前诊断中的应用.中华检验医学杂志,2016,39:613⁃617.]
[19]Wang F,Han L,Yang Y,Gu X,Ye J,Qiu W,Zhang H,Zhang Y,GaoX,WangY.Clinical,biochemical,and molecular analysis of combined methylmalonic acidemia and hyperhomocysteinemia(cblC type)in China.J Inherit Metab Dis,2010,33 Suppl 3:435⁃442.
[20]Huemer M,Diodato D,Schwahn B,Schiff M,Bandeira A,Benoist JF,Burlina A,Cerone R,Couce ML,Garcia⁃Cazorla A,la Marca G,Pasquini E,Vilarinho L,Weisfeld⁃Adams JD,Kožich V,Blom H,BaumgartnerMR,Dionisi ⁃ViciC.Guidelines for diagnosis and management of the cobalamin⁃related remethylation disorders cblC,cblD,cblE,cblF,cblG,cblJ and MTHFR deficiency.J Inherit Metab Dis,2017,40:21⁃48.
[21]Fraser JL,Venditti CP.Methylmalonic and propionic acidemias:clinicalmanagement update.Curr Opin Pediatr,2016,28:682⁃693.
Analysis of clinical phenotype and genetic mutation with outcome evaluation in one family of vitamin B12⁃dependent methylmalonic aciduria
LI Jing1,SUN Yi⁃ming2,OU Li⁃yu1,ZHU Yu⁃ling1,WANG Liang1,LI Huan1,ZHANG Cheng11Department of Neurology,2Department of Health Care Clinic,the First Affiliated Hospital,Sun Yat⁃sen University,Guangzhou 510080,Guangdong,China
LI Jing and SUN Yi⁃ming contributed equally to this study
ZHANG Cheng(Email:zhangch6@mail.sysu.edu.cn)
ObjectiveTo explore the clinicalfeatures,genetic mutation and vitamin B12therapeutive effectiveness in vitamin B12⁃dependent methylmalonic acidemia(MMA).MethodsClinical data in a pedigree of 4 members with vitamin B12⁃dependent MMA was collected.Peripheral blood samples were collected for plasma amino acids and acylcarnitines and gene muatation analysis.The therapeutic efficacy ofvitamin B12was evaluated.ResultsThe initialpresentations ofthe proband were underachievement and personality changes in 12⁃year old,and accompanied by visual hallucinations and weakness of lower limbs during the course of disease.The younger brother of the proband presented with bad⁃temper and lower acheivement.The analysis of plasma amino acid and acylcarnitine showed that proband and his younger brother's plasma propionylcarnitine and propionylcarnitine/acetylcarnitine ratio,and the level of methylmalonic acid in urine were increased significantly. Compound heterozygeous mutation of c.482G>A(p.Arg161Gln)and c.609G>A(p.Trp203X)in MMACHC gene were seen in the proband and her younger brother.Her father carried MMACHC gene missense mutation c.482G>A(p.Arg161Gln),while her mother carried MMACHC gene nonsense mutation c.609G>A(p.Trp203X).Symptoms of the proband were improved after vitamin B12therapy.ConclusionsThe late⁃onset vitamin B12⁃dependent MMA is caused by compound heterozygote mutation of MMACHC gene.It had good responsive to vitamin B12therapy.Early diagnosis and timely treatment may play a critical role for the outcomes of patients with this disease.
Methylmalonic acid; Metabolism,inborn errors; Vitamin B12; Phenotype;Genes; Mutation; Pedigree
10.3969/j.issn.1672⁃6731.2017.07.009
国家自然科学基金资助项目(项目编号:81471280);广东省广州市2015年产学研专项项目(项目编号:1561000153);国家自然科学基金青年科学基金资助项目(项目编号:81601087);广东省科学技术厅2014年度公益研究与能力建设专项资金资助项目(项目编号:2014A020212130);国家自然科学基金资助项目(项目编号:81271401);国家自然科学基金-广东省联合基金重点资助项目(项目编号:U1032004)
利婧、孙毅明并列为本文第一作者
510080广州,中山大学附属第一医院神经科[利婧、欧俐羽(现在广西中医药大学附属瑞康医院神经内科,邮政编码:530011)、朱瑜龄、王倞、李欢、张成],保健科(孙毅明)
张成(Email:zhangch6@mail.sysu.edu.cn)
This study was supported by the National Natural Science Foundation of China(No.81471280,81271401),2015 Production,Study and Research Special Project of Guangzhou,Guangdong Province,China(No.1561000153),the National Natural Science Foundation of China for Young Scientists(No.81601087),Non⁃Profit Study and Capability Building Special Fund Support Project of Guangdong Provincial Department of Science and Technology,China in the Year 2014(No.2014A020212130),and Joint Fund of National Natural Science Foundation of China and Natural Science Foundation of Guangdong Province,China(No.U1032004).
2017⁃06⁃12)
