头部震颤伴小脑萎缩一例临床表型及基因突变分析
2017-11-21谢坤铭顾卫红郝莹陈园园张瑾张鑫
谢坤铭 顾卫红 郝莹 陈园园 张瑾 张鑫
头部震颤伴小脑萎缩一例临床表型及基因突变分析
谢坤铭 顾卫红 郝莹 陈园园 张瑾 张鑫
目的通过对1例头部震颤伴小脑萎缩患者临床表型和基因检测结果进行综合分析,明确诊断疾病并探讨基因检测结果的解读方法.方法与结果采集1例30岁男性患者临床表型,进行二代基因测序和Sanger测序验证,通过中文人类表型标准用语、基因检索工具Phenomizer、Ensembl数据库、在线人类孟德尔遗传数据库相关信息,对基因检测结果进行解读.结果显示,患者存在脊髓小脑共济失调19型(SCA19型)致病基因KCND3基因杂合突变c.1057A>G(p.Ser353Gly),其父母均未携带该突变基因;患者还存在帕金森病20型致病基因SYNJ1基因杂合突变c.4436C>T(p.Thr1479Ile),其母携带该突变基因.表型相似度分析显示,患者表型与SCA19型一致,KCND3基因变异位点c.1057A>G在不同物种同源基因中具有高度保守性.结论通过对患者临床表型和基因检测结果综合分析,KCND3基因杂合突变c.1057A>G(p.Ser353Gly)为致病性突变.
脊髓小脑共济失调; 表型; 基因; 突变
共济失调作为常见的神经系统疾病表现,涉及 多种病因,包括单基因遗传病、散发性神经变性病和获得性疾病.从遗传学角度看,神经系统遗传性疾病约占所有遗传性疾病的60%,遗传性共济失调占神经系统遗传性疾病的10%~15%,迄今已报道100余种类型[1].遗传模式可以分为常染色体显性遗传(AD)、常染色体隐性遗传(AR)、X连锁遗传和线粒体母系遗传,具有高度临床异质性和遗传异质性,其中,脊髓小脑共济失调(SCA)呈常染色体显性遗传[2⁃3],临床主要表现为平衡障碍、肢体协调障碍、步态不稳、构音障碍和眼球运动障碍等,且伴复杂神经系统损害,如锥体系、锥体外系、视觉、听觉、脊髓和周围神经系统损害等,主要病理损害部位为小脑、脑干和脊髓.目前报道的亚型达40余种[4⁃5].本文详细分析1例头部震颤伴小脑萎缩患者的临床表型并对其基因检测结果进行深入解读.
病例资料
一、临床特征
患者 男性,30岁,因头颈部不自主晃动2年,于2013年11月6日至中日友好医院运动障碍与神经遗传病专病门诊就诊.患者2年前无明显诱因出现头颈部不自主晃动,发作时间持续1~2 min或数天不等,肩部晃动僵硬,紧张和精细动作时易发作,进行性加重;言语模糊,构音不清,偶有吞咽困难,睡眠时好时坏,可见胸闷、呼吸困难、反复发作的神经性皮炎.既往史和个人史无特殊.父母身体健康,非近亲婚配,无明确家族性遗传性疾病病史.门诊神经系统检查:神志清楚,轻度构音障碍;眼球各向活动不充分,外展露白,未见眼震;头部阵发性晃动;四肢肌力5级,双上肢肌张力降低,腱反射减弱,双下肢肌张力正常,腱反射正常,足跖反射正常,病理反射未引出;双侧指鼻试验、跟⁃膝⁃胫试验稳准.简易智能状态检查量表(MMSE)评分正常.实验室检查各项指标均于正常值范围.影像学检查:头部MRI显示小脑重度萎缩,脑干中度萎缩(图1).神经电生理学检查:心电图、肌电图和脑电图均未见明显异常.结合临床表现和家族史,临床拟诊为常染色体隐性遗传性共济失调.予金刚烷胺50 mg/次、2次/d,苯海索(安坦)1 mg/次、2次/d,氯硝西泮1 mg/d口服治疗.至2016年3月复诊时头部晃动明显缓解.
二、基因检测
分别抽取患者及其父母外周静脉血5 ml,送检北京金准基因科技有限责任公司,进行3994种人类孟德尔遗传性疾病致病基因外显子二代基因测序和家系验证.对候选变异位点采用Sanger测序进行家系验证,提示患者存在2种可能的致病性突变:(1)KCND3基因杂合突变c.1057A>G(p.Ser353Gly).Sanger测序显示其父母均未携带该突变基因(图2).KCND3基因与SCA19型相关,呈常染色体显性遗传,理论上1条染色体突变即可致病.(2)SYNJ1基因杂合突变c.4436C>T(p.Thr1479Ile).Sanger测序证实此突变来自其母(图3).SYNJ1基因与帕金森病20型(PARK20型)相关,呈常染色体隐性遗传,理论上必须2条等位染色体同时出现致病性突变方可致病(即纯合突变或复合杂合突变致病).

图1 头部MRI检查显示,小脑呈现重度萎缩(粗箭头所示),脑干呈现中度萎缩(细箭头所示)1a 横断面T1WI 1b 矢状位T2WIFigure1 Head MRIshowed severecerebellaratrophy(thick arrows indicate)and moderate brain stem atrophy(thin arrow indicates). AxialT1WI(Panel1a). SagittalT2WI(Panel 1b).
三、突变的致病性分析
突变的致病性分析步骤为:(1)将核心表型转化为中文人类表型标准用语/人类表型标准用语(CHPO/HPO,http://www.chinahpo.org/).(2)候选基因筛选,根据对应的CHPO/HPO词汇,采用基因检索工具Phenomizer(http://compbio.charite.de/phenomizer/)检索候选基因.(3)根据美国医学遗传学和基因组学会(ACMG)制定的变异位点分级指南(http://wintervar.wglab.org/),输入突变基因信息,结合核心表型、家族史和遗传模式,即可判断突变基因的致病性."Pathogenic"表示已知致病性突变,"likely pathogenic"表示疑似致病性突变,"VUS"表示临床意义不明的突变,"likely benign"表示疑似良性突变,"benign"表示良性突变.(4)保守性分析,通过 Ensembl数据库(http://useast.ensembl.org/index.html)获取多物种相关基因序列,采用BioEdit软件(http://www.bioedit.com/)比对变异位点在不同物种的序列保守性.(5)表型相似度分析,通过检索在线人类孟德尔遗传数据库(http://www.omim.org/)比对疾病表型与患者表型.
1.核心表型转化为中文人类表型标准用语 采用CHPO搜索引擎检索本文患者的核心表型,结果参见表1.


图2 该例患者及其父母KCND3基因Sanger测序所见 2a 患者存在KCND3基因c.1057A>G(p.Ser353Gly)杂合突变(红色圆圈所示)2b 其父不携带KCND3基因突变(红色圆圈所示)2c其母不携带KCND3基因突变(红色圆圈所示)Figure 2 The Sanger sequencing of KCND3 gene of the patient and his parents.Heterozygous mutation of KCND3 gene c.1057A>G(p.Ser353Gly)in the patient(red circle indicates,Panel 2a).No KCND3 gene mutation in the patient's father(red circle indicates,Panel 2b)and mother(red circle indicates,Panel 2c).


图3 该例患者及其父母SYNJ1基因Sanger测序所见 3a 患者存在SYNJ1基因c.4436C>T(p.Thr1479Ile)杂合突变(红色圆圈所示) 3b 其父不携带SYNJ1基因突变(红色圆圈所示)3c 其母存在SYNJ1基因c.4436C>T(p.Thr1479Ile)杂合突变(红色圆圈所示)Figure 3 The Sanger sequencing of SYNJ1 gene of the patient and his parents.Heterozygous mutation of SYNJ1 gene c.4436C>T(p.Thr1479Ile)in the patient(red circle indicates,Panel 3a).No SYNJ1 gene mutation in the patient's father(red circle indicates,Panel 3b).Heterozygous mutation of SYNJ1 gene c.4436C>T(p.Thr1479Ile)in the patient's mother(red circle indicates,Panel 3c).
2.基于人类表型标准用语筛选候选基因 通过基因检索工具Phenomizer,筛选致病基因,结果显示,相关度最大的基因为KCND3基因(P=0.000).
3.确定致病性突变 根据美国医学遗传学和基因组学会制定的变异位点分级指南,KCND3基因为"pathogenic",即致病性突变;SYNJ1基因为"likely pathogenic",即疑似致病性突变.
4.保守性分析 通过Ensemble数据库检索KCND3基因及其他物种同源基因序列,采用BioEdit软件进行比对分析,结果显示,变异位点在不同物种的进化中具有高度保守性(图4).
5.表型相似度分析 通过在线人类孟德尔遗传数据库检索KCND3和SYNJ1基因相关疾病表型,与患者表型进行比对.核心表型为小脑萎缩、头颈部不自主晃动、腱反射减弱、构音障碍和吞咽困难,与KCND3基因的疾病表型(SCA19型)高度相似,提示为致病基因(表2).SYNJ1基因的疾病表型为PARK20型,表现为面具脸、目光凝视、眼睑失用症、扫视障碍、核上性凝视麻痹、下颌震颤、动作迟缓、肌僵直、拖步、姿势不稳、震颤、肌张力障碍、认知功能障碍和早发型癫发作,头部MRI显示大脑皮质萎缩[6⁃7],与患者表型不符,排除SYNJ1基因致病的可能.经基因检测最终明确诊断为SCA19型,继续予药物治疗以缓解相应症状.
讨 论
脊髓小脑共济失调呈常染色体显性遗传,自1993年SCA1型致病基因定位以来,根据研究者对其致病基因定位的时间顺序,由国际人类基因组组织基因命名委员会(HGNC)命名,至2017年5月已定位43个致病基因位点,其中28种致病基因已被克隆(http://neuromuscular.wustl.edu/ataxia/domatax.html).脊髓小脑共济失调家系中大部分为致病基因动态突变,采用毛细管电泳片段分析法可检出.随着二代基因测序技术的发展,越来越多的非动态突变逐渐被发现,其致病性尚待进一步明确.
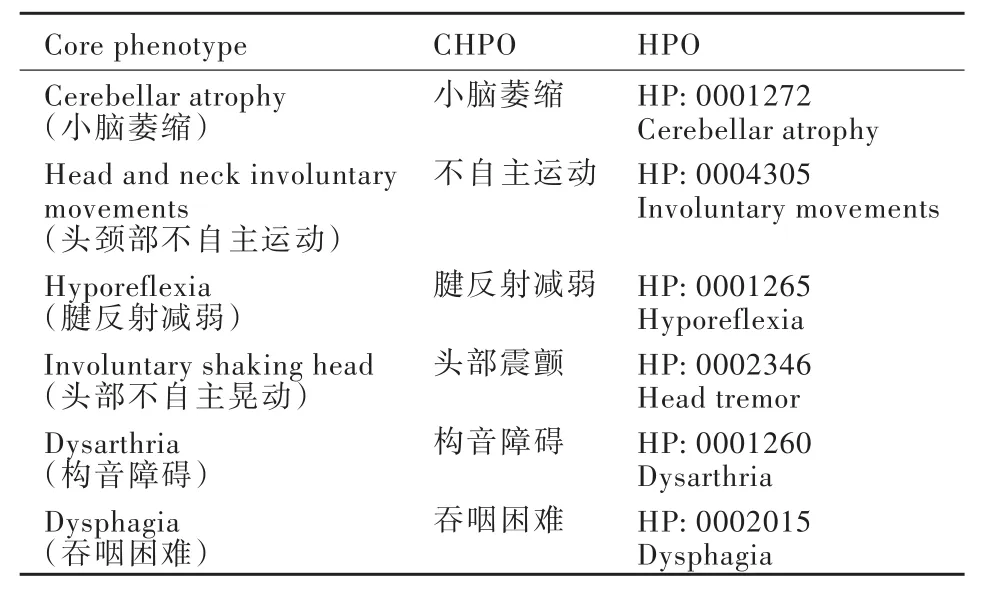
表1 该例患者核心表型及其对应的CHPO/HPOTable 1. The core phenotype and corresponding CHPO/HPO of the patient

图4 保守性分析显示,KCND3基因变异位点c.1057A>G在不同物种同源基因中具有高度保守性Figure 4 Conservation analysis showed the variation locus of KCND3 gene c.1057A>G was highly conservative with the homologous gene in different species.
该例患者28岁起病,无明确家族史,最初考虑为常染色体隐性遗传性共济失调,通过二代基因测序并结合临床表型分析,检出SCA19型相关基因KCND3基因杂合突变c.1057A>G(p.Ser353Gly),为新发突变,且在进化过程中高度保守.
SCA19型发病年龄多样化,自青年至中年均可发病,进展缓慢.已报道的家系多有小脑萎缩症状,以头部晃动发病,伴轻至中度构音障碍,其他症状还包括凝视诱发眼震,吞咽困难,小脑、步态、躯干、肢体共济失调,腱反射减弱,姿势性震颤,肌阵挛,认知功能障碍和踝振动觉受损等[8].SCA19型最早发现于一荷兰家系,先证者约31岁时以头部晃动发病,继而出现构音障碍,逐渐出现行走困难,神经系统查体显示不规则震颤,频率缓慢,姿势变化和站立时头颈部不自主晃动,细小眼震,眼球各向活动充分,关节位置觉受损、双足振动觉减退,四肢协调性受到严重影响,表现出辨距不良和快复轮替动作笨拙,共济失调步态[9].Verbeek等[10]通过全基因组测序(WGS)将致病基因定位于1p21~q21.2003年一中国脊髓小脑共济失调家系致病基因定位于1p21~q23,当时被国际人类基因组组织基因命名委员会命名为SCA22型[11].但在2004年,Schelhaas等[12]发现SCA19型和SCA22型的基因突变位点相重合,至此认为二者为脊髓小脑共济失调的同一亚型.2012年,Duarri等[13]的研究显示,KCND3基因为SCA19型致病基因.
KCND3蛋白系编码电压门控性钾离子通道(VGKC)的Shal家族α亚单位,对细胞膜复极化具有重要作用[14],包括6个跨膜片段和细胞内氨基末端(N末端)和羧基末端(C末端).逆转录⁃聚合酶链反应(RT⁃PCR)和测序分析证实1个剪切位点突变KCND3L,插入编码1个含19个氨基酸的多肽链,包含1个磷酸化位点,其中较短亚型为KCND3S[15].Northern blotting法检测显示,长度为8.50X103bp的转录本在脑和心脏呈高表达,而在肾脏、肝脏、肺、胰腺、脾或骨骼肌中未检测到[16].这种长转录本主要见于丘脑、尾状核、白质和骨骺,短转录本主要见于额叶、枕叶和小脑皮质[17].1999年首次在人类大脑和心脏中克隆出KCND3基因,RT⁃PCR法发现仅KCND3基因长片段在心脏表达[18],包含7个外显子,长度约25X 103bp,而较短亚型由6个外显子编码.KCND3基因突变导致其编码的电压门控性钾离子通道特性改变,引起智力障碍、癫发作、注意力缺陷、多动、斜视和口唇失用等症状[19].
该例患者头颈部不自主晃动,肩部晃动僵硬,紧张和精细动作时易发作,进行性加重,言语模糊;神经系统检查,轻度构音障碍,眼球各向活动不充分,未见眼震,阵发性头部晃动,四肢肌力5级,双上肢肌张力偏低,腱反射减弱,双下肢肌张力正常,腱反射适中,足跖反射正常,双侧指鼻试验、跟⁃膝⁃胫试验稳准;头部MRI显示明显小脑萎缩,上述表型与荷兰SCA19型家系临床表型相似.
SYNJ1基因相关疾病表型为PARK20型,临床特点为运动迟缓、肌肉僵直、静止性震颤、姿势不稳、核上性凝视麻痹、痴呆、肌张力障碍和全面性强直⁃阵挛发作(GTCS)[20],左旋多巴或其他拟多巴胺类药治疗有效[21].该例患者帕金森病症状并不明显,且该突变位点源自其母,其父无突变,该基因为常染色体隐性遗传,因此考虑SYNJ1基因致病的可能性不大.

表2 该例患者KCND3基因相关疾病表型与患者表型相似度分析Table 2. Phenotypic similarity analysis of the patient
在对该例患者临床表型和基因检测结果进行综合分析解读过程中,我们采用CHPO/HPO,有助于表型标准化并连接基因数据库进行解读.HPO旨在提供人类疾病中用于描述表型异常的标准词汇,从医学文献、Orphanet数据库、DECIPHER数据库和在线人类孟德尔遗传数据库获得的信息进行开发,目前包含11 896个名词,同时还提供一套针对约4000种疾病的注释.HPO一直持续进行词条维护和完善.目前国内大部分医疗和科研工作者所使用的表型描述词汇并无统一标准,大多数医院信息系统中记录的临床表型数据均为非结构化的自然词汇,给人类疾病尤其是遗传性疾病的临床研究和交流带来不便,对后期数据挖掘和分析也造成很大困扰,因此,在中国建立一套标准的临床表型词汇非常重要.CHPO作为开放平台,旨在联合各相关领域专业人士,逐步建立中文临床表型词汇标准,并指导和服务于中文使用者的临床和科研工作,目前已建立搜索引擎http://www.chinahpo.org/.
本研究通过对1例无明确家族史的头部震颤伴小脑萎缩患者的临床表型和基因数据进行综合分析,探讨表型标准化结合分析软件工具进行基因检测选点和结果解读的方法和流程:(1)将患者核心表型转化为CHPO/HPO.(2)将患者的HPO结果输入基因检索工具,如Phenomizer,筛选可能的候选致病基因.(3)将二代基因测序检出的基因变异位点与上述分析结果相结合进行选点.(4)根据美国医学遗传学和基因组学会制定的变异位点分级指南进行致病性评价.(5)表型相似度分析,基于在线人类孟德尔遗传数据库记录的临床表型与患者表型进行细致比对.随着二代基因测序技术的快速发展,基因检测费用下降,在高通量测序获得大量变异选点过程中,表型数据与基因数据的有效连接至关重要.整个流程涉及多个环节,由不同专业背景的人员和机构参与,如何确保信息的可靠性,以及在传递过程中避免信息丢失和发生偏倚,需建立共同语言.HPO即为很好的连接方式,可以将医师所了解的患者表型信息、检测机构获得的患者基因信息、疾病和基因数据库三者进行连接,为遗传性疾病的诊断提供重要支持工具.
[1]Liu ZL,Liang XL,Zhang C.Neurogenetics.3rd.Beijing:People's Medical Publishing House,2011:85[.刘焯霖,梁秀龄,张成.神经遗传病学.3版.北京:人民卫生出版社,2011:85.]
[2]Matilla⁃Dueñas A,Sánchez I,Corral⁃Juan M,Dávalos A,Alvarez R,Latorre P.Cellular and molecular pathways triggering neurodegeneration in the spinocerebellar ataxia.Cerebellum,2010,9:148⁃166.
[3]Chen JW,Zhao L,Zhang F,Li L,Gu YH,Zhou JY,Zhang H,Meng M,Zhang KH,Le WD,Dong CB.Clinical characteristics,radiological features and gene mutation in 10 Chinese families with spinocerebellar ataxias.Chin Med J(Engl),2015,128:1714⁃1723.
[4]Serrano⁃Munuera C,Corral⁃Juan M,Stevanin G,San Nicolás H,Roig C,Corral J,Campos B,de Jorge L,Morcillo⁃Suárez C,Navarro A,Forlani S,Durr A,Kulisevsky J,Brice A,Sánchez I,Volpini V,Matilla⁃Dueñas A.New subtype of spinocerebellar ataxia with altered vertical eye movements mapping to chromosome.JAMA Neurol,2013,70:764⁃771.
[5]Tsoi H,Yu AC,Chen ZS,Ng NK,Chan AY,Yuen LY,Abrigo JM,Tsang SY,Tsui SK,Tong TM,Lo IF,Lam ST,Mok VC,Wong LK,Ngo JC,Lau KF,Chan TF,Chan HY.A novel missense mutation in CCDC88C activates JNK pathway and causes a dominant form of spinocerebellar ataxia.J Med Genet,2014,51:590⁃595.
[6]Chen KH,Wu RM,Lin HI,Tai CH,Lin CH.Mutational analysis of SYNJ1 gene(PARK20)in Parkinson's disease in a Taiwanese population.Neurobiol Aging,2015,36:2905.
[7]Kirola L,Behari M,Shishir C,Thelma BK.Identification of a novel homozygous mutation Arg459Pro in SYNJ1 gene of an Indian family with autosomal recessive juvenile parkinsonism.Parkinson Relat Disord,2016,31:124⁃128.
[8]Schelhaas HJ,van de Warrenburg BP.Clinical,psychological,andgenetic characteristicsofspinocerebellarataxia type 19(SCA19).Cerebellum,2005,4:51⁃54.
[9]Schelhaas HJ,Ippel PF,Hageman G,Sinke RJ,van der Laan EN,Beemer FA.Clinical and genetic analysis of a four⁃generation family with a distinct autosomal dominant cerebellar ataxia.J Neurol,2001,248:113⁃120.
[10]Verbeek DS,Schelhaas JH,Ippel EF,Beemer FA,Pearson PL,Sinke RJ.Identification of a novel SCA locus(SCA19)in a Dutch autosomal dominant cerebellar ataxia family on chromosome region 1p21⁃q21.Hum Genet,2002,111:388⁃393.
[11]Chung MY,Lu YC,Cheng NC,Soong BW.A novel autosomal dominant spinocerebellar ataxia(SCA22)linked to chromosome 1p21⁃q23.Brain,2003,126:1293⁃1299.
[12]Schelhaas HJ,Verbeek DS,Van de Warrenburg BP,Sinke RJ.SCA19 and SCA22:evidence for one locus with a worldwide distribution.Brain,2004,127:E6.
[13]Duarri A,Jezierska J,Fokkens M,Meijer M,Schelhaas HJ,den Dunnen WF,van Dijk F,Verschuuren⁃Bemelmans C,Hageman G,van de Vlies P,Küsters B,van de Warrenburg BP,Kremer B,Wijmenga C,Sinke RJ,Swertz MA,Kampinga HH,Boddeke E,Verbeek DS.Mutations in potassium channel KCND3 cause spinocerebellar ataxia type 19.Ann Neurol,2012,72:870⁃880.
[14]Lee YC,Durr A,Majczenko K,Huang YH,Liu YC,Lien CC,Tsai PC,Ichikawa Y,Goto J,Monin ML,Li JZ,Chung MY,Mundwiller E,Shakkottai V,Liu TT,Tesson C,Lu YC,Brice A,Tsuji S,Burmeister M,Stevanin G,Soong BW.Mutations in KCND3 cause spinocerebellar ataxia type 22.Ann Neurol,2012,72:859⁃869.
[15]Duarri A,Lin MC,Fokkens MR,Meijer M,Smeets CJ,Nibbeling EA,Boddeke E,Sinke RJ,Kampinga HH,Papazian DM,Verbeek DS. Spinocerebellar ataxia type 19/22 mutations alter heterocomplex Kv4.3 channel function and gating in a dominant manner.Cell Mol Life Sci,2015,72:3387⁃3399.
[16]Kong W,Po S,Yamagishi T,Ashen MD,Stetten G,Tomaselli GF.Isolation and characterization of the human gene encoding Ito:further diversity by alternative mRNA splicing.Am J Physiol,1998,275:H1963⁃1970.
[17]Isbrandt D,Leicher T,Waldschütz R,Zhu X,Luhmann U,Michel U,Sauter K,Pongs O.Gene structures and expression profiles of three human KCND(Kv4)potassium channels mediating A⁃type currentsⅠ(TO)andⅠ(SA).Genomics,2000,64:144⁃154.
[18]Dilks D,Ling HP,Cockett M,Sokol P,Numann R.Cloning and expression of the human kv4.3 potassium channel.J Neurophysiol,1999,81:1974⁃1977.
[19]Duarri A,Nibbeling E,Fokkens MR,Meijer M,Boddeke E,Lagrange E,Stevanin G,Brice A,Durr A,Verbeek DS.Erratum to:the L450F corrected mutation in KCND3 brings spinocerebellar ataxia and Brugada syndrome closer together.Neurogenetics,2015,16:243.
[20]De Rosa A,Pellegrino T,Pappatà S,Lieto M,Bonifati V,Palma V,Topa A,Santoro L,Bilo L,Cuocolo A,De Michele G.Non⁃motor symptoms and cardiac innervation in SYNJ1⁃related parkinsonism.Parkinsonism Relat Disord,2016,23:102⁃105.
[21]QuadriM,Fang M,Picillo M,OlgiatiS,Breedveld GJ,Graafland J,Wu B,Xu F,Erro R,Amboni M,Pappatà S,Quarantelli M,Annesi G,Quattrone A,Chien HF,Barbosa ER;The International Parkinsonism Genetics Network;Oostra BA,Barone P,Wang J,Bonifati V.Mutation in the SYNJ1 gene associated with autosomal recessive,early⁃onset parkinsonism.Hum Mutat,2013,34:1208⁃1215.
Clinical phenotype and genetic mutation of one case with head tremor and cerebellar atrophy
XIE Kun⁃ming1,GU Wei⁃hong2,HAO Ying2,CHEN Yuan⁃yuan2,ZHANG Jin2,ZHANG Xin21Grade 2014,College of Traditional Chinese Medicine,Beijing University of Chinese Medicine,Beijing 100029,China
2Movement Disorder&Neurogenetics Research Center,China⁃Japan Friendship Hospital,Beijing 100029,China Corresponding author:GU Wei⁃hong(Email:jane55.gu@vip.sina.com)
ObjectiveTo make the diagnosis for a patient presented with head tremor and cerebellar atrophy by integrating clinical features and accessory examination with genetic testing and to explore the interpretation of genetic testing results.MethodsA 30⁃year⁃old male patient's medical information,clinical pheontype,family history and accessory examinations were collected.The next⁃generation sequencing(NGS)of exons in 3994 causative genes of Mendelian inheritance diseases and the family tree verification were carried out.China Human Phenotype Ontology(CHPO),Phenomizer,Ensembl and Online Mendelian Inheritance in Man(OMIM)database were used to interpret the genetic test results.ResultsThe patient carried heterozygous mutation of spinocerebellar ataxia type 19(SCA19)related KCND3 gene c.1057A>G(p.Ser353Gly),but his parents did not carry this mutation.The patient also carried heterozygous mutation of parkinsonism type 20(PARK20)related SYNJ1 gene c.4436C>T(p.Thr1479Ile)which was also seen in his mother.Phenotypic similarity analysis showed the patient's phenotype was correspond with the phenotype of SCA19,and the variation locus of KCND3 gene c.1057A>G was highly conservative with homologous gene in different species.ConclusionsBy means of the integration of clinical phenotype with the result of genetic test,KCND3 gene c.1057A>G(p.Ser353Gly)carried in the patient is the pathogenic mutation.
Spinocerebellar ataxias; Phenotype; Genes; Mutation
This study was supported by Grant Awarded 2010-2012 from Ministry of Health Foundation of China.
10.3969/j.issn.1672⁃6731.2017.07.007
卫生部部署(管)医院2010-2012年度临床学科重点项目
100029北京中医药大学中医学院2014级(谢坤铭);100029北京,中日友好医院运动障碍与神经遗传病研究中心(顾卫红,郝莹,陈园园,张瑾,张鑫)
顾卫红(Email:jane55.gu@vip.sina.com)
2017⁃06⁃08)
