介电层表面直接生长石墨烯的研究进展∗
2017-11-10杨慧慧高峰戴明金胡平安
杨慧慧 高峰 戴明金 胡平安
1)(哈尔滨工业大学材料科学与工程学院,哈尔滨 150080)
2)(哈尔滨工业大学,微系统与微结构制造教育部重点实验室,哈尔滨 150080)
介电层表面直接生长石墨烯的研究进展∗
杨慧慧1)2)高峰1)2)戴明金1)2)胡平安1)2)†
1)(哈尔滨工业大学材料科学与工程学院,哈尔滨 150080)
2)(哈尔滨工业大学,微系统与微结构制造教育部重点实验室,哈尔滨 150080)
(2017年8月5日收到;2017年9月26日收到修改稿)
石墨烯,介电层,化学气相沉积,无转移
1 引 言
石墨烯是一种由碳原子构成的二维层状纳米材料.其中碳原子以sp2轨道杂化方式成键,组成六边形基本结构单元,并按照蜂窝状在二维空间内排列.晶格常数C—C键长1.42 Å,单层石墨烯厚度3.35 Å.自2004年Geim等[1]通过机械剥离的方法得到单层石墨烯以来,关于石墨烯的研究层出不穷,这主要是由于其突出的物理、化学性质与巨大的应用价值.单层石墨烯为一种零带隙的半金属性材料,其色散关系曲线为线性锥形,导带与价带在布里渊区顶点处相交.石墨烯的载流子有效质量接近于零,其在电场下的迁移率可达到105cm2·V−1·s−1, 远远高于传统硅基材料的载流子迁移率.随着研究的不断深入,科学家们在石墨烯体系中发现了诸多奇异的物理现象,如:低温下反常量子霍尔效应[2],室温下的量子霍尔效应[3]、亚微米的弹道输运距离[4]、Klein隧穿[5]等,极大地推动了凝聚态量子物理的研究进展.与此同时,石墨烯兼具超高的比表面积、优异的导热性、超高的力学性质与可见透光性,因而在柔性场效应晶体管以及光电器件、生化传感与催化、超级电容器、能源存储等领域均具有广阔的应用前景.
为了满足科学研究与工业应用的需求,制备大面积、高质量的石墨烯具有十分重要的意义.人们发展出了多种制备石墨烯的方法,如微机械剥离法、化学剥离法、化学氧化还原法、化学气相沉积(CVD)法等.其中,CVD法由于具有生长石墨烯可控性高、结晶质量好、均匀、薄膜尺寸大等优势而成为制备石墨烯最为普遍的方法之一.这种方法大多以过渡金属为生长基底,借助于其较高的化学催化活性,促进碳源裂解为CHx碎片小分子和C原子,并在金属表面吸附、扩散、成核、生长形成石墨烯.通过调控生长过程的参数,可以实现大面积、层数可控、高质量且结构均一连续的石墨烯薄膜.经过不断的工艺优化,可以实现超大面积的石墨烯单晶的生长(>1 cm)[6,7].然而在实际应用的过程中,在金属表面形成的石墨烯一般需要通过有机聚合物(聚甲基丙烯酸甲酯,PMMA)辅助法将其转移至介电层上,才可以进行下一步的微电子元器件组装.而在转移过程中,1)PMMA无法完全去除,少量聚合物会残留在石墨烯表面,对石墨烯形成掺杂或形成载流子陷阱[8−10];2)在刻蚀过程中易引入金属杂质,同样会对石墨烯的结构与性质产生不利影响[11];3)湿法转移过程中会给石墨烯引入额外的H2O与O2,从而产生掺杂效应[12];4)转移后石墨烯表面存在许多褶皱、裂纹以及破损,破坏了薄膜的连续性[13].总之,聚合物辅助转移过程对石墨烯的质量造成了不可避免的损伤,严重降低了其电学性能,同时复杂的转移过程也为石墨烯器件的制备增加了成本.
为解决这一问题,研究者将目光投向在介电衬底表面直接生长石墨烯.一般来说,在缺乏金属催化的情况下,石墨烯需要在较高的温度下才可以形成.介电层表面能量较低,对碳源小分子的裂解以及石墨烯的形成所产生的催化作用十分微弱[14],因而在介电衬底上直接生长石墨烯是一个研究难点.面对这一挑战,研究者改进了石墨烯的生长手段,并尝试在多种介电衬底(Al2O3,h-BN,Si3N4,玻璃等)上生长石墨烯,目前已经取得了显著成果.对这一研究方向已有较为深入的综述[14,15],但仍需要更加系统的分析与整理.本文综述了近年来在介电基底上直接生长石墨烯的研究进展,总结了在介电层上直接合成石墨烯的不同生长方法,并对所生长石墨烯的质量进行了评估与比较.
2 生长方法
为实现石墨烯在介电基底表面的直接生长,人们探索并发展出多种生长方法与策略,同时系统地探究了石墨烯的生长过程,并对所生长的石墨烯的质量进行了分析.其生长方法可以大致分为金属辅助法、等离子体增强法以及热力学或动力学调控法,下文中将对此进行具体的阐述.
2.1 金属辅助法
为了实现在介电层表面直接生长石墨烯,在研究初期,人们首先充分研究了过渡金属表面生长石墨烯的机理.该过程主要可分为两种生长机理:表面催化机理与溶解扩散机理.高温下前驱体首先裂解为小分子及原子,以Cu等在生长温度区间内对于碳的溶解度可忽略不计的金属为生长基底时,碳原子首先吸附在金属表面并聚集成核形成石墨烯岛,在其表面迁移的C原子或团簇在成核点处与边缘原子连接并不断扩展生成石墨烯,即表面催化机理.另一类生长机理以Ni,Co等具有高溶碳性的金属基底为代表.C原子溶解进入金属表面内部,并在降温过程中从体相中偏析——沉淀,在表面成核生长.这类金属表面形成的石墨烯多为双层甚至多层[16].
根据石墨烯的生长机理,研究者创新性地采用金属辅助催化生长工艺以直接生长石墨烯.其中一种思路便是利用金属的高催化活性,将厚度为几百纳米的金属薄膜沉积到生长基底上构成夹层结构,催化石墨烯在金属薄膜表面或者金属与介电层之间的界面处形成(如图1(a)所示).该金属薄膜可以在生长后通过化学方法去除,通过控制参数也可以实现在生长过程中将金属薄膜直接蒸发[17−24].从这种方法延伸出来,可以利用气态的金属原子作为催化剂,从而实现在介电层上直接生长石墨稀[25−29].另一种思路便是利用碳在Ni等金属中具有较高的溶解度以及在其内部较大的扩散、迁移的能力,制作碳源/金属薄膜/介电基底三明治结构,通过加热退火的方式加速碳原子的渗透与石墨烯的形成,令其在介电衬底与金属的界面处形成石墨烯[30−34].
2.1.1 金属薄膜辅助催化生长
Levendorf等[17]率先利用金属薄膜取代传统的金属箔片作为催化剂,在二氧化硅表面沉积一层金属薄膜(5 nm Ni/500 nm Cu),以甲烷为前驱体,在Cu薄膜表面生长获得单层石墨烯,如图1(b)所示.采用这种方法,他们制备了尺寸大于1 cm的单层石墨烯薄膜,并成功地在介电层上直接制备了石墨烯晶体管阵列,该石墨烯的迁移率达到了700 cm2·V−1·s−1.Ismach等[18]采用类似的方法,利用金属的去湿润以及蒸发过程,在生长石墨烯的同时,铜薄膜不断地蒸发并减少,最终在介电层直接得到石墨烯薄膜(如图1(c)所示),而避免了后续的化学刻蚀过程.但这种方法制备的石墨烯表面铜残留较明显,且缺陷、褶皱较多,薄膜的连续性也有待提高.

图1 (a)金属薄膜辅助生长石墨烯的过程示意图;(b)石墨烯器件的光学图,插图为石墨烯沟道不同位置处的拉曼谱图[17];(c1)利用金属薄膜催化石墨烯生长2 h并原位蒸发的样品的光学图片;(c2),(c3)依次为石墨烯拉曼I(2D),I(G)/I(2D)的面扫图[18]Fig.1.(a)Schematic of the fabrication process by metal assisted growth of graphene on dielectric layers;(b)optical microscope image of the graphene device,and inset is the Raman spectra of the graphene at different places alone the channel[17];(c1)optical microscopeimage of the sample after 2h simultaneous growth of graphene and evaporation of Cu;Raman mapping of(c2)I(2D),(c3)I(G)/I(2D)[18].
当生长石墨烯的前驱物为固态的自组装材料(SAM)、无定形碳(a-C)、聚合物等时,金属薄膜辅助生长法同样适用,并且固态前驱物的分子种类、浓度等因素对石墨烯产物的质量均有影响.Shin等[19]分别采用三种自组装SAM单层碳材料(分别为辛基、十八烷基、苯基有机硅烷)为生长原料,将其处于SiO2基底与金属Ni薄膜中间并制备了大面积的石墨烯.研究发现,所生长石墨烯的层数与SAM分子中的碳原子个数直接相关.以脂肪族SAM为前驱物时,生长的石墨烯薄膜表面存在许多空洞以及岛状结构;以芳香族SAM为前驱物时,生长的石墨烯表面形貌则更加均匀.前驱物的构型对所形成石墨烯的质量影响较大,Zhuo等[23]的报道也得出了类似的结论.采用相似的结构,Yan等[20]尝试了包括SAM在内的多种固态碳源作为前驱物生长石墨烯,如苯丙基聚甲基硅氧烷、PMMA、聚苯乙烯、丙烯腈-丁二烯-苯乙烯共聚物聚合物,石墨烯的层数主要通过改变前驱体的浓度进行控制.在优化的生长条件下,可以在多种基底上实现双层石墨烯的生长.但是这种方法仍然需要后续的化学刻蚀过程以除掉上层覆盖的Ni.为改进这一过程,Xiong等[22]利用直流磁控溅射技术,分别在多种介电层上依次沉积5 nm厚的a-C薄膜和65 nm的金属Ni薄膜.在1100°C的条件下快速退火,表面的Ni完全蒸发,介电层上形成了高质量的石墨烯.如图2所示,石墨烯的拉曼光谱显示石墨烯具有较高的质量,D峰较低.对于400—800 nm可见光,其透光率高于94%.经过分析发现,Ni的快速蒸发与a-C的存在有着直接的关系.在退火过程中,形成了亚稳相Ni3C,其不断分解与蒸发.因而该过程中石墨烯可能是从Ni3C相中析出并形成的.同时X射线光电子能谱(XPS)测试发现少量的Ni渗透进入石墨烯内部形成了掺杂.这种方法制备的石墨烯层数可控,可生长4 in(1 in=2.54 cm)的大面积薄膜.其薄层方阻低至50 Ω·sq−1,如此低的方阻值可能是由于石墨烯中存在一定程度的Ni原子的掺杂所造成的.

图2 (a)在熔融石英、蓝宝石、石英以及二氧化硅/硅片上表面生长晶圆级石墨烯的光学图片;(b),(c)Ni/C图案以及相应的石墨烯图案;(d),(e)石墨烯薄膜的拉曼谱图与可见透射光谱图[22]Fig.2.(a)Optical images of the original substrates,Ni/C,and the grown graphene on fused silica,sapphire,quartz and SiO2/Si;optical microscope images of(b)the Ni/C pattern and(c)graphene pattern on SiO2/Si;(d)Raman spectrum and(e)transmittance spectrum of the graphene film[22].
利用金属薄膜辅助催化生长石墨烯时,可以在介电层表面直接获得大面积连续的石墨烯薄膜,实现了直接生长的目的.但金属的掺杂与残留无法完全避免,石墨烯的结晶质量与电学性能仍有待进一步提高.
2.1.2 气态金属原子辅助催化生长
在以沉积的金属薄膜作为石墨烯生长的催化剂时,表面的金属残留仍然十分明显,并直接影响到生成石墨烯的晶体质量.受到气态二茂铁催化生长碳纳米管的启发,Teng等[25]利用气态Cu以及H原子催化前驱物的分解与石墨烯的形成.在常压条件下,1000°C时铜的平衡蒸气压为3×10−7bar(1 bar=105Pa),此时体系中存在许多气态Cu原子与H原子.如图3(a)所示,这些气态催化剂在与甲烷分子碰撞的过程中令甲烷分子裂解为碳原子或自由基,随着载气输送至下游的SiO2/Si基底上,并形成连续的石墨烯薄膜.另一种可能的生长机理是气态中的Cu原子沉积在二氧化硅表面形成Cu纳米粒子,催化石墨烯在二氧化硅表面形成,并随着反应的进行,Cu纳米粒子二次蒸发,从而实现了在介电层基底上直接生长石墨烯.该石墨烯电子的迁移率在100—600 cm2·V−1·s−1范围内.
然而从该报道中的拉曼谱图中可以看到,石墨烯D峰强度显著,I2D/IG分布不均一,说明生成的石墨烯晶体缺陷较大,晶粒尺寸较小,并且薄膜均匀性较差,层数难以控制.这可能是由于在二氧化硅表面石墨烯的成核过程随机发生,并且C原子在其表面的迁移能力较差造成的.另外一种可能则是由于体系中气态Cu原子不充足而导致甲烷分解不完全引起的.Choi小组[26]对这一结构进行了优化,采用图3(b)中的生长方法,将铜箔置于介电层正上方,但不与介电层基底直接接触.提高了介电层上方Cu蒸气的密度,并通过优化气态Cu/CH4的比例,得到了缺陷少、均匀性好的单层石墨烯(>95%),与在Cu箔表面生长的石墨烯质量相当.XPS测试分析显示反应过程中无铜硅化合物以及碳硅化合物的存在,并且反应后二氧化硅表面不存在任何形式的Cu元素残留.该石墨烯构筑的场效应晶体管(FET)空穴以及电子的迁移率分别为800 cm2·V−1·s−1和700 cm2·V−1·s−1.利用相同的方法,Liu小组[29]在石英基底上生长可以得到大面积均匀的石墨烯薄膜,制备的石墨烯具有较高的质量,透过率为82%时,薄层电阻达到370—510 Ω·sq−1,并且可以直接应用于触摸控制板器件中,无需任何中间转移过程.
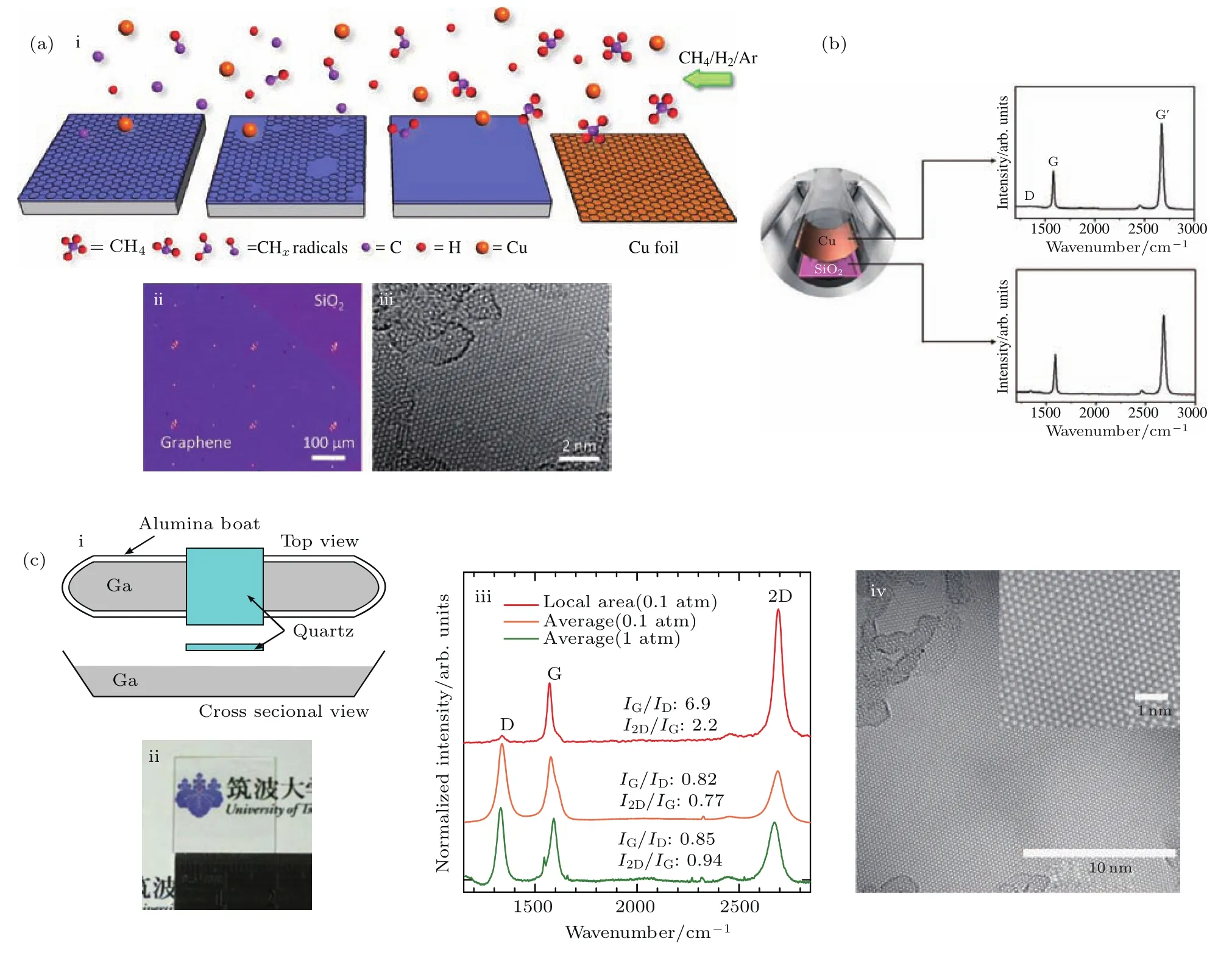
图3 (a-i)为气态Cu原子催化生长石墨烯的示意图,(a-ii)为在二氧化硅表面生长石墨烯薄膜的光学图片,(a-iii)为采用种子法生长的石墨烯薄膜的透射电子显微镜(TEM)图[25];(b)将Cu片置于生长基底上方催化生长石墨烯的示意图,以及生长在Cu表面以及介电衬底表面的石墨烯的拉曼谱图[26];(c-i)为气态Ga原子催化生长石墨烯的生长示意图,(c-ii)为所生长石墨烯的光学图,(c-iii)为石英表面石墨烯薄膜的拉曼谱图,(c-iv)为石墨烯薄膜的高分辨透射TEM(HRTEM)图像[28]Fig.3.(a-i)Illustration of gas phase catalysis of the growth of graphene on SiO2/Si,(a-ii)transferred graphene onto SiO2/Si,(a-iii)TEM image of the prepared graphene[25];(b)setup of the growth procedure and the Raman spectra of the graphene films on Cufoil and on SiO2[26];(c-i)setup of the graphene growth procedure by Ga vapor catalysis,(c-ii)photograph of the graphene film on quartz,(c-iii)Raman spectra of the as-grown graphene films,(c-iv)HRTEM image of the graphene[28].
研究表明,利用这种方法制备的石墨烯的质量和面积与体系中气态金属原子的密度有着直接的联系.尽管相较于铜,金属镍的熔点较高,1000°C时平衡蒸气压较小.但Yen等[27]利用气态Ni原子辅助催化,在950—1100°C范围内成功实现了石墨烯的生长.通过调控生长温度可以控制所生长石墨烯的层数.实验证明在Ga,In等金属对石墨烯的生长具有良好的催化效果[35],在这些液态金属的表面也可以实现石墨烯的生长[36].这类金属熔点相对较低,将其作为气态催化剂,可以提高体系中漂浮的金属蒸气的含量,加强对石墨烯的催化效果.如金属Ga的熔点极低,1000°C时平衡蒸气压比Cu高出一个数量级,同时高温下对于C原子的溶解性较低.Murakami等[28]采用图3(c)中的生长方法,将石英片置于装有Ga单质的瓷舟内,以气态Ga原子为催化剂,在二氧化硅上实现了2 mm×2 mm石墨烯薄膜的生长.暗场TEM图片揭示了石墨烯薄膜大部分由单层构成,晶粒尺寸大小在50—200 nm范围内.该方法生长得到的石墨烯薄膜局部为单层结构,D峰十分微弱,2D峰强度突出.但大部分位置为多层结构,I(D)/I(G)与I(G)/I(2D)值较大.证明该石墨烯内部晶粒尺寸较小,晶界处缺陷较多,散射现象比较严重.这也解释了其迁移率偏低(<10 cm2·V−1·s−1) 的原因.
金属蒸气既可以发挥催化石墨烯生长的作用,又不会在介电衬底表面引入过多的金属残留.相较于其他气态金属,气态Cu具有较为合适的蒸气压与催化活性.在优化的实验条件下生长得到的石墨烯薄膜与在铜箔表面生长的石墨烯薄膜质量相当.这种方法有望成为工业制备石墨烯的工艺之一.
2.1.3 固态扩散-析出反应
以上在介电层上生长石墨烯的手段主要是利用过渡金属的催化性,而对于Ni等溶碳性较强的金属而言,可以利用C原子在金属内部溶解、扩散、析出的原理,制作介电层基底/金属/碳源的夹层结构,在加热退火的过程中,前驱物在金属表面分解并溶解至金属体相内部,通过晶界处或晶粒内部逐渐扩散,并源源不断地输送至金属与介电层之间,在界面处成核、生长,形成连续的石墨烯薄膜.
Kong小组[30]以及Tour小组[31]采用相近的方法,在介电层基底上沉积一定厚度的Ni薄膜,以固态或气态碳源为前驱物,在1000°C左右的高温下,前驱物裂解为C原子并充分溶解在Ni薄膜层内,在内部扩散迁移.降温过程中C原子不仅在金属Ni-气态界面析出,同时也在金属Ni-介电层基底界面析出成核并生长石墨烯.利用这种迁移析出的方法得到的石墨烯薄膜连续性良好,但均匀性一般.该薄膜中多为单层以及双层结构,同时也存在多层石墨烯.Kwon小组[34]发现在低于260°C甚至在室温条件下可以通过C在Ni中的扩散迁移,在Ni/SiO2界面处形成石墨烯.文中提出,C原子在金属Ni内部的扩散分为两种,一种为晶界扩散,另一种为晶体内扩散.在较低的温度(≤260°C)下,C在晶界处的扩散迁移速率要远远大于在晶粒内部的迁移速率.如图4(a)所示,C原子首先通过晶粒边界处迁移到达金属与基地的界面处,成核生长并形成多层的石墨烯桥,C原子持续不断地扩散到金属与基底界面处,沿着石墨烯桥的边缘平面生长,得到双层以及单层的石墨烯.在塑料或者玻璃基底上也可以通过同样的操作得到石墨烯,晶粒尺寸平均为40—50 nm.这种方法制备得到的石墨烯晶粒尺寸与Ni薄膜中晶粒尺寸大小相当.为提高石墨烯晶粒尺寸,可以对沉积在SiO2/Si基底上的Ni薄膜进行高温退火处理,提高其晶粒尺寸,可以相应地增大制备的石墨烯晶粒尺寸至15—20µm.但是这种方法制备的石墨烯薄膜层数无法保证其均一性,并且晶粒尺寸仍然有很大的提高空间.
Kato等[33]将等离体子技术与这种扩散-析出的方法结合起来,在合适的Ni层厚度(55 nm)与快速的生长时间(≤5 min)共同作用下,可以实现石墨烯的选择性生长,即只在金属Ni与SiO2/Si界面之间生长石墨烯,而在Ni表面没有石墨烯生成.如图4(b)所示,采用这种生长结构,可以直接制备图案化的石墨烯,其拉曼面扫图以及拉曼谱图显示,生长的石墨烯具有较好的质量与连续性.
利用Ni原子在多晶金刚石中快速扩散的性质,可以在金刚石表面形成单层至多层的石墨烯[37].研究者们在多晶金刚石表面沉积了一层金属Ni,在高温退火的过程中,Ni原子迅速扩散至金刚石内部,表面痕量Ni原子作为石墨烯的成核点,金刚石内部的C原子不断扩散,可在表面形成晶圆级的石墨烯薄膜.
在金属Cu中碳原子的溶解性相对较小,在其内部的迁移能力远远弱于在金属Ni内部的迁移能力.在一些报道中,相同的条件下,以Cu作为扩散金属层时,无法得到目标产物.这可能正是由于C原子在Cu中扩散迁移能力相对较弱所导致的[31].但Su等[32]在合适的工艺窗口条件下,实现了C原子在Cu内部扩散迁移并在Cu与SiO2/Si或石英等生长基底的界面之间形成连续的石墨烯薄膜.他们发现,过薄的Cu层厚度,过高的生长温度或者过低的生长压力均会导致Cu的显著蒸发与去湿.而薄膜厚度超过300 nm或生长温度较低时,C原子在Cu内部从表面扩散到界面处较为困难,因而难以获得连续性良好的石墨烯,甚至根本无法形成石墨烯.在优化条件下制备的石墨烯的载流子迁移率为670 cm2·V−1·s−1,狄拉克点远远偏离0 V(>100 V),说明石墨烯样品为重度P掺杂.
金属辅助法生长石墨烯有效避免了传统的转移过程,保留了金属的高催化活性,成功实现了在介电衬底上制备连续的石墨烯薄膜.生长温度与在Cu箔上生长石墨烯的温度相当(1000°C),甚至更低(260°C).在保留金属催化作用的同时,又与传统的薄膜制作工艺相容,具有十分广大的应用空间.
2.2 等离子体增强CVD(PECVD)法
PECVD是一种利用耦合产生具有高化学活性的等离子体,发生化学反应并沉积在生长基底表面上的一种薄膜制备技术,主要是借助强电场或磁场令反应气体电离产生等离子体.这种等离体子包括分子、原子、自由基、离子以及大量自由电子等组成,这些活性基团化学性质活跃,易吸附到生长基底表面,初步形成晶核,并逐步生长、扩展为平面薄膜.这种技术在保留普通CVD方法制备薄膜材料的可控性高,可制备大面积连续薄膜的特点的同时,降低了反应所需要的温度,加快了薄膜生长的速率.
Zhang课题组首先在550°C的条件下利用远程PECVD法,在硅片、石英、玻璃等表面上实现了石墨烯薄膜的生长[38].扫描隧道显微镜(STM)分析显示,所制备的石墨烯薄膜是由平面尺寸为十几纳米、层数为2—4层的石墨烯纳米晶所组成.中国台湾清华大学的Chiu课题组[39]利用电子回旋共振化学气相沉积法制备了连续的石墨烯薄膜,该石墨烯薄膜晶粒尺寸为3—5 nm,由混合的单层、双层以及多层构成.然而利用这种方法生长的石墨烯晶粒尺寸较小,且层数不可控,甚至为纳米团簇或纳米晶结构.Wei等[40]对PECVD系统在介电层上生长石墨烯的过程进行了系统的研究.他们发现,通过控制等离子体中H2的刻蚀与碳源的生长这两种过程的平衡,可以实现石墨烯的可控刻蚀与生长.首先他们以剥离的三层石墨烯薄层作为研究对象,研究了在PECVD生长过程中不同生长参数对于该石墨烯样品的影响.研究结果表明在600°C,90 mTorr(1 Torr=1.33322×102Pa),30%H2等离子体气氛下生长60 min时,C原子在石墨烯薄片的边缘处平面生长.当生长温度相对较低时,或者氢气浓度相对较高时,刻蚀作用占主导地位.当生长温度较高或氢气浓度较低时,在石墨烯薄层表面以及二氧化硅表面均易形成团簇.这种现象可以利用以下化学式进行解释:

式中Q为反应释放的热量.控制石墨烯生长与刻蚀之间的平衡过程,在成核与生长的临界温度左右时,可以实现在石墨烯边缘C原子继续生长.图5中具体列出了石墨烯生长的临界条件.探究了这两种平衡过程后,他们分别采用了剥离的石墨烯薄层、PECVD沉积的石墨烯纳米团簇与图案化刻蚀的石墨烯样品作为石墨烯晶体生长的种子,首先利用氢气等离子体活化,刻蚀掉石墨烯种子边缘存在的一些缺陷结构,露出裸露的具有高活性的原子,继而在适合的临界生长条件下实现石墨烯的平面生长.其中以石墨烯纳米团簇作为生长种子时,可以得到结晶性良好的,晶粒尺寸为1—2µm的单层六边形石墨烯.该石墨烯样品的迁移率在550—1600 cm2·V−1·s−1范围内. 当以NH3与CH4作为前驱物时,可以利用这种直接生长n型掺杂的石墨烯薄膜[41].

图5 (a)利用临界PECVD法生长石墨烯的过程示意图;(b)不同压力下,生长温度、H2含量对石墨烯生长结果的关系曲线;(c)生长石墨烯不同阶段的原子力显微镜(AFM)图像[40]Fig.5.(a)Schematic diagram of the graphene growth by critical PECVD method;(b)plots of the results of the graphene growth as a function of temperature and H2content under different pressure;(c)AFM images of the graphene at different growth stages[40].
一般情况下,石墨烯生长温度为900—1050°C.几乎没有电子器件可以承受这种程度的高温处理而不被破坏,因而将石墨烯引入电子器件,均需要借助于中间转移步骤,而难以实现直接将石墨烯嵌入器件结构中.利用PECVD方法可以大幅降低石墨烯的生长温度至600°C,从而实现在发光二极管(LED)器件结构上的直接生长,同时还可保持LED活性层的有序结构[42].
另外,在制备图案化二维石墨烯的过程中,一般采用在完成生长转移光刻这一流程构造预期的图案化结构;另一种制作方法是首先制备图案化的金属基底,在此基底上生长转移得到预期结构.这两种制作流程均不可避免地涉及到材料的转移以及基底的化学刻蚀,而利用PECVD方法,将掩膜板直接覆盖在基底表面,即可以在介电层上制备得到图案化结构的石墨烯[43].
一般介电层表面能很低,催化活性较小,在其表面直接生长石墨烯化学势垒较高.PECVD作为一种有效克服高势垒化学反应的生长手段,可以在较低的温度下实现高质量石墨烯的成核以及生长.同时相较于高温反应,PECVD对于介电衬底的热稳性要求较低,对其表面破坏较小.相较于其他方法,PECVD制备图案化的石墨烯薄膜具有简单直接、不引入中间化学过程的优点,在集成化电子器件等应用中具有突出的优越性.
2.3 热力学或动力学调控法
2.3.1 氧辅助生长——热力学调控
在介电层表面生长石墨烯的主要难点在于成核与生长过程缓慢,这主要是由于在没有金属催化剂的情况下,石墨烯成核与生长的化学驱动力较低.引入一些中间反应或过渡态,可以为石墨烯的生长提供一条热力学上更为有利的反应路径,使其更加容易发生.
在探索二维材料生长工艺的过程中,研究者发现氧气的引入可以有效地改善材料的生长结果[44],其根本原因在于高温下O原子与C原子、H原子具有较强的键合作用,改变了石墨烯成核与生长的热力学路径,降低了反应所需要的势垒高度.对SiO2/Si表面在800°C条件下在空气气氛中进行热处理,随后在1100°C生长3 h,可以得到单层的石墨烯岛.延长生长时间至8 h,石墨烯岛相互扩展合并成为连续的多层石墨烯薄膜[45].这可能是由于体系中少量的氧气通过C—O键与H—O键的结合增强了其对CHx碎片的俘获能力,提高C—C偶联以及成核的概率,从而促进了石墨烯的直接生长.XPS分析以及拉曼谱图均显示其形成了良好的sp2结构,其载流子迁移率达到了531 cm2·V−1·s−1.研究发现,氧气的存在同样可以促进在介电层表面直接生长二维h-BN薄膜[46].这说明,可以通过向体系中引入O原子以调控二维材料生长的热力学过程,从而制备出满足实际需要的高质量的样品.
2.3.2 近平衡态生长——动力学调控
在过渡金属表面生长石墨烯的实验研究中,人们发现通过降低反应前躯体的浓度或者降低反应速率,可以实现在近似平衡的条件下生长二维材料.通过改变动力学参数可以调控石墨烯生长的过程,从而优化石墨烯生长的质量.Liu等[47]发现,降低碳源的流量,同时延长生长时间,可以在Si3N4、石英、蓝宝石等基底表面实现较大石墨烯单晶的生长.在该生长条件下,反应近似接近平衡态,进入反应体系的C原子逐一与石墨烯边缘原子结合以保证体系能量最低.该石墨烯单晶具有规则的六边形以及十二边形形貌,单晶尺寸最大达到11µm.Rümmeli课题组[48]采用了一种新奇而又简单的方法生长得到了高质量且大面积的单层石墨烯.其生长示意图见图6(a).在其他条件相同的情况下,当二氧化硅层正面朝上生长时,所得到的石墨烯薄膜大部分为单层结构,局部区域为多层结构.将二氧化硅层翻转过来,倒扣在Al2O3基底上时,所得到的石墨烯表面仍随机分布一些多层的结构,但这种多层石墨烯的比例明显减少.而将两片SiO2/Si垂直堆叠在一起,得到的石墨烯则为均匀的单层结构.图6(b)显示了这三种不同的结构所生长的石墨烯的扫描电子显微镜(SEM)图像.由于二氧化硅层表面十分平滑,粗糙度仅为0.18 nm±0.02 nm.因而在这种夹层结构中,上下平行的表面构造了相对局限的空间.理论分析表明,在双层硅片之间形成了高度为1.4 nm的限制空间.在该空间内,前驱物分子的供应速率被降低,其扩散速率同样受到了限制,但仍足以保证其稳定流经二氧化硅表面反应区.在这种动力学情况下,石墨烯的成核密度得到了显著的降低,生长得到均匀的单层石墨烯.拉曼以及AFM测试均表明,在这种结构上,表面形成了严格的单层结构,并且石墨烯质量较高,缺陷较少.生长1 h时,表面形成了375 nm±30 nm的单层石墨烯岛.进一步延长生长时间至5 h,孤立的单层石墨烯岛相互融合成为连续薄膜,并且没有出现其表面继续生长第二层或第三层石墨烯的现象.该石墨烯薄膜为严格的单层结构.该石墨烯在550 nm处透光率为97%,载流子迁移率达到410—760 cm2·V−1·s−1. 这种限制空间的方法极大地提高了生长石墨烯的质量与可控性.

图6 (a)限制空间法生长石墨烯示意图;(b)采用二氧化硅层正面朝上、二氧化硅层正面朝下以及两层二氧化硅层相对的结构生长的石墨烯的SEM图像[48]Fig.6.(a)Setup of the sandwich configuration for graphene growth;(b)SEM images of the graphene grown by three configurations:facing up,facing down and sandwich[48].
3 生长基底
在非金属衬底表面生长石墨烯,其生长过程与生长基底具有十分紧密的联系.在不同的生长基底表面,石墨烯的生长模式、生长机理与产品质量均有较大的差别.介电基底的晶格结构、介电性质、高温稳定性等对石墨烯的生长与器件的性能会产生显著的影响.下文将聚焦于在不同的生长基底表面生长石墨烯的过程及结果,并对其进行分析与总结.
3.1 氧化物类介电基底(SiO2/Si,Al2O3以及SrTiO3)
SiO2/Si为常见的介电衬底,广泛应用于微电子器件与电路中.表面SiO2层的介电常数为3.9,介电强度为106—107V·cm−1.SiO2/Si与目前主流的硅电子工艺完全兼容,在许多器件结构如金属-氧化物-半导体、金属-绝缘层-半导体结构中均为重要的结构单元,尤其是作为栅极材料而被广泛应用于FET中.目前二维材料的生长以及电学性质研究也多以SiO2/Si为基础展开.在SiO2/Si基底上直接生长石墨烯已有大量的报道,在前文中已经提到,利用金属辅助催化、PECVD等方法,均可实现在SiO2/Si表面生长石墨烯薄膜,并且可以直接制作图案化的石墨烯,直接应用于集成电路与器件中,此处不再赘述.
然而该石墨烯薄膜的迁移率与在Cu箔上生长的石墨烯的迁移率相比,相差较多(多为500 cm2·V−1·s−1左右).这可能是由于二氧化硅表面的电荷转移、吸附、电子声子散射等原因导致石墨烯的电学性能下降.另一方面,研究发现,在生长石墨烯的温度(1000°C)下,表面的SiO2介电性质会受到一定的影响,降低了其表面石墨烯的电学性能.因而在低温下生长石墨烯或在其他高温下稳定的介电衬底上生长石墨烯具有重要的意义.
采用具有超高热稳定性和高介电常数的栅极介电材料作为基底,既可满足高温条件下在介电层上生长石墨烯的条件,又可以有效避免栅极电流的泄漏,增加晶体管电容,屏蔽表面散射效应,改善器件的载流子迁移率.Al2O3及SrTiO3均为常见的高介电材料,探究在其表面直接生长石墨烯具有十分重要的研究价值与应用空间.
蓝宝石(α-Al2O3)是一种六方晶格结构的原子晶体.Al原子与O原子以共价键方式结合,其熔点高达2045°C.常用的切面为a面,c面以及r面,其(0001)晶面参数为a=4.758 Å,c=12.991 Å.完美的Al2O3介电常数为9,具有较强的击穿强度(1 MV·cm−1)与较小的泄漏电流.美国康奈尔大学的Hwang等[49]率先在1350—1650°C条件下以丙烷为碳源,在c面(0001)蓝宝石单晶表面生长得到了层数为9—100层的石墨烯薄膜.Fanton等[50]研究了1425—1600°C范围内在蓝宝石表面石墨烯的生长情况(如图7(a)所示).研究发现,随着生长温度的提高,石墨烯的I(D)/I(G)的比值逐渐下降(低至0.05),表明其结晶质量逐渐提高.在1525°C的条件下生长的石墨烯薄膜,其中>90%的面积为单层结构.该石墨烯薄膜在较长距离内粗糙度小于1 nm,载流子迁移率大于3000 cm2·V−1·s−1.Hwang等[51]也探究了甲烷-蓝宝石体系生长石墨烯的工艺条件.研究发现,采用较高的甲烷浓度(>0.6%)时,则可以在较宽的温度窗口内通过一步法直接得到石墨烯.在他们的实验中,最佳的生长条件为1550°C,甲烷浓度为1.2%,H2/CH4比例为12.在较低的甲烷浓度(<0.2%)以及较高的生长温度(>1450°C)下,C原子的刻蚀过程反应速率大于其形成石墨烯的反应速率,无法在蓝宝石表面形成石墨烯.因而采用较低的甲烷浓度(<0.2%)时,需要在相对较低的温度(<1350°C)下成核,在高温(1450—1650°C)下平面生长的两步法来实现石墨烯的生长.图8(b)中的拉曼谱图表明,这种一步法(高C浓度)以及两步法(低C浓度)均可以在蓝宝石表面得到质量较高的石墨稀.明显不同的是,两步法制备的石墨烯具有更高的载流子密度,并且体现出p型掺杂效应.这可能是由于两步法生长过程中蓝宝石表面O原子的掺杂所造成的.该石墨烯晶粒尺寸约270 nm,载流子迁移率可达到300—2050 cm2·V−1·s−1.
在蓝宝石表面生长石墨烯时,成核以及生长温度越高,得到的石墨烯质量越好.然而较高的温度(约1550°C)显然增加了生长石墨烯的成本,也提高了对仪器的要求.为进一步降低石墨烯在蓝宝石基底上的生长温度,Miyasake等[52]改用乙醇为碳源,将石墨烯在a面(110)蓝宝石基底上的生长温度降低至800—1000°C.然而这种方法得到的石墨烯的2D峰强度比较微弱.石墨烯单晶的尺寸小于15 nm.浦项科技大学的 Choi组[53]探索了以甲烷为前驱体,950°C的温度下在蓝宝石表面生长石墨烯.该石墨烯2D峰强度有所增强,但D峰也十分明显.
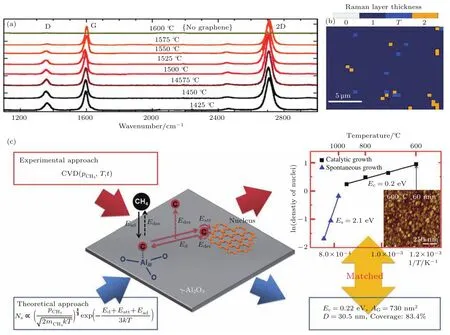
图7 (a)在1425—1600°C温度范围内生长的石墨烯的拉曼谱图;(b)展示了在1525°C生长的石墨烯的层数拟合,其中>90%为单层石墨烯[50];(c)石墨烯在γ-Al2O3表面生长的机理示意图[54]Fig.7.(a)Raman spectra of the graphene films grown on sapphire at temperature from 1425 °C to 1600 °C;(b) fi tted layer number of the graphene film,monolayer coverage>90%[50];(c)growth model of the growth of graphene on γ-Al2O3[54].
韩国科学技术研究院的Song小组[54]探究了γ-Al2O3的催化石墨烯生长的性能.以甲烷作为碳源,在600—1050°C条件下,在γ-AL2O3表面生长得到了覆盖率较高的石墨烯薄膜.同时,他们探究了γ-Al2O3催化生长石墨烯的机理,如图7(c)所示.石墨烯在γ-Al2O3表面生长的过程中,由于γ-Al2O3对于C的溶解度极小,因而生长遵循与Cu类似的表面吸附原理.该过程包括:表面吸附、表面扩散、原子连接过程.它们所对应的能量势垒依次为Ead,Ed,Eatt.其中表面吸附过程控制着C原子的供应速率;扩散过程决定了石墨烯产品的均一性;原子连接过程决定着石墨烯的平面生长质量.通过下列公式可以模拟计算出石墨烯生长的反应活化能的理论值,Ec约为0.22 eV,Es约为2.09 eV,该结果与实验的结果完全符合.

Liu课题组[55]尝试了在一种高介电常数材料SrTiO3(001)基底上生长石墨烯,在1000°C,生长3 h左右时,在其表面直接得到了连续的单层石墨烯薄膜.该石墨烯在550 nm处透光率为96.4%, 薄层方阻达到950 Ω·sq−1.FET测试其转移曲线表明,在低至0.11 V的偏压下,该石墨烯显示出双极性特征,载流子迁移率达到了870—1050 cm2·V−1·s−1.
在实际应用中,介电层的介电强度、泄漏电流等性质对于微电子器件的性能有着重要的影响.Al2O3,SrTiO3等均是常见的高介电常数材料,在其表面生长的石墨烯往往表现出更加优异的电学性质.除此之外,在ZnO[56],MgO[57]等氧化物基底表面也可以实现石墨烯的生长.在不同的介电衬底上生长石墨烯可以满足不同的实际需要,为石墨烯的应用拓宽了基础.
3.2 六方氮化硼
近年来,二维六方氮化硼(h-BN)逐渐吸引了研究者的广泛研究.一方面这是由于h-BN具有5.9 eV的带隙结构,介电常数为3—4,介电强度为8 MV·cm−1[58],作为二维电子材料器件中的介电层基底具有显著的优势.另一方面h-BN化学性质稳定,具有较强的抗高温、抗氧化性以及良好的导热性,具有极大的应用前景.
考虑到h-BN的晶格参数(a=2.504 Å)与石墨烯的晶格参数(a=2.468 Å)相近,晶格失配仅为1.7%,并且以h-BN作为生长基底外延生长石墨烯可以形成周期性势场,对石墨烯的能带结构产生影响,这对于调控石墨烯的能带结构,开发石墨烯的潜在功能具有十分重要的意义.Xie团队[59]首先在剥离的h-BN上生长得到了多层的石墨烯.在后续的研究报道中,该小组在h-BN表面生长得到了尺寸最大约为270 nm的石墨烯单晶,并且发现石墨烯优先在螺旋位错以及台阶结构处成核生长[60].同时,Choi等[61]与Mishra等[62]均在无催化剂的条件下在h-BN表面生长得到了圆形的单层石墨烯片.Zhang课题组[63]利用PECVD技术在剥离的h-BN上制备了高质量的石墨烯单晶(如图8所示).每个单晶的晶体取向均一致,证明该石墨烯是由底层h-BN经过范德瓦耳斯外延生长得到.AFM测试显示,底层h-BN与石墨烯形成了有序的摩尔条纹,周期波长为15 nm±1 nm,由此可证明h-BN与石墨烯的晶格取向严格一致,旋转角度为0°.该值与理论计算的结果[64]和其他报道中的结果[65]完全一致.在这种严格的范德瓦耳斯外延生长模式下,当相近的两个晶粒扩大合并时,连接处原子结构仍有序排列,并无晶粒边界存在.因而这种方法生长的石墨烯可以合成制备高质量并且大面积的单晶石墨烯结构,由该石墨烯/h-BN异质结制备的FET器件显示其载流子迁移率在1.5 K时可达到约5000 cm2·V−1·s−1.

图8 (a)h-BN表面外延生长石墨烯的示意图;(b)石墨烯/h-BN的拉曼谱图;(c)h-BN表面外延生长石墨烯的AFM图像[63]Fig.8.(a)Sketch of the epitaxial growth of graphene on h-BN;(b)Raman spectra of graphene/h-BN sheets;(c)AFM image of the epitaxial graphene grown on h-BN[63].
然而,由于剥离的h-BN平面尺寸一般为若干微米,基于这种h-BN基底生长石墨烯,无法得到大面积的连续的薄膜.因而Wang等[66]将目光转向CVD方法制备的大面积的h-BN薄膜.在铜箔上首先生长h-BN薄膜,继而生长石墨烯单层薄膜,相较于利用CVD法制备并转移至h-BN或SiO2/Si基底上的石墨烯,该石墨烯具有更高的载流子迁移率,狄拉克点更加接近0 V.这是由于直接在h-BN上生长石墨烯,界面更加清洁,减少了杂质与陷阱的引入,因而体现出更加接近于本征石墨烯的电子输运性质.Liu课题组[67]发现,以苯甲酸作为生长石墨烯的碳源,在h-BN/Cu表面可以通过调节温度来实现平面或垂直石墨烯/h-BN的生长.其基本原理在图9(a)中给出:>950°C时,苯甲酸分解产生的CO2与h-BN发生反应,将h-BN刻蚀掉一部分,因而倾向于形成平面结构;<950°C时,刻蚀反应不发生,此时倾向于生长垂直结构.
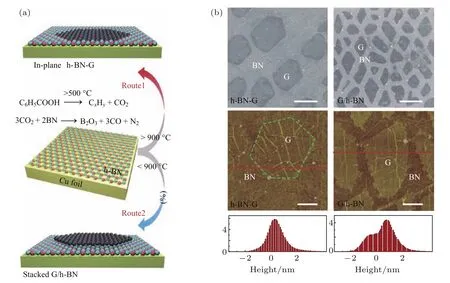
图9 (a)温度调控石墨烯选择性平面/垂直生长示意图;(b)平面、垂直石墨烯/h-BN异质结SEM图像以及AFM图像[67]Fig.9.(a)Schematic of the temperature-triggered selective of lateral and vertical growth of graphene on h-BN;(b)SEM images,AFM images and height pro fi les of the lateral and vertical heterostructures[67].
研究发现,无论是在剥离的h-BN还是在Cu箔表面生长的h-BN,将其作为基底生长石墨烯时,生长速率均十分缓慢,晶粒尺寸难以增大.这是由于h-BN具有较低的催化活性的原因.为提高其生长速率与晶粒尺寸,可以在生长过程中引入气态催化剂.如将Ge或硅烷加入到石墨烯的生长体系中,在1280°C时,在剥离的h-BN表面石墨烯的生长速率由原来的5 nm·min−1分别提高至50 nm·min−1,400 nm·min−1,并且晶粒尺寸大幅提高[68].以二茂镍作为碳源时,气态Ni原子与Cu基底的协同催化作用,石墨烯的生长速率会得到大幅提高.相较于以苯甲酸为碳源生长石墨烯,其生长速率提高了8—10倍,最大单晶尺寸可达20µm[69].
h-BN具有优异的介电性与稳定性,以其作为介电层可以大幅提高石墨烯等二维材料的器件性能.并且在与底层h-BN形成周期排列时,石墨烯的能带结构以及量子性质均可能发生改变,如二级狄拉克点的衍生、室温下的量子振荡等现象.因而在h-BN表面制备高质量的石墨烯具有十分重要的研究价值与应用空间.目前,在h-BN表面制备得到的石墨烯具有结晶质量高、单晶尺寸大、生长速率快、界面清洁、性能突出的优势.
3.3 SiC以及Si3N4
SiC具有多种同质多象变体.常见的有2H,3C,4H,6H,15R等.其禁带宽度约3 eV,介电常数为10左右,击穿强度为4 MV·cm−1.在探索石墨烯生长的初级阶段,单晶SiC高温外延法便用来制备得到了高质量的单层石墨烯薄膜.在真空或者常压条件下,在大于1200°C的条件下,单晶SiC(0001)Si终止或(000)C终止表面Si原子在高温下脱除,剩余表面C原子重构,形成石墨烯结构[70,71].这种方法制备的石墨烯质量较高,结晶性好,迁移率可达到104cm2·V−1·s−1,并且与Si半导体工艺完全兼容.
然而经过理论分析以及实验研究发现,在SiC表面外延生长石墨烯的过程中,首先在SiC表面形成一层C原子构成的缓冲层,该缓冲层中的C原子以共价键的形式与SiC基底结合具有较大的带隙宽度.该缓冲层上形成的第二层C薄膜才开始体现出石墨烯的色散关系[72],如图10(a)所示.该缓冲层的存在严重制约了SiC(0001)表面外延生长石墨烯的电学性质,因而如何消除缓冲层成为了研究的难点.Virojanadara等[73]利用图10(b)中可逆加氢反应的原理,成功将缓冲层直接转化为石墨烯.在对SiC/缓冲层C/石墨烯结构在室温下进行氢气等离子体处理,或在高温氢原子气氛下处理,活性H离子或原子会透过缓冲层,与SiC基底顶层Si原子成键,从而破坏缓冲层C原子与Si原子的共价键,使得缓冲层转变为sp2结构的石墨烯.这种方法直接得到了双层石墨烯结构,避免了缓冲层的存在.除了插入氢原子外,一些其他非金属原子如F[74],O[75]以及金属原子Li[76],Si[77],Ge[78]等也可以与顶层Si键合,消除缓冲层,使其退耦与上层石墨烯构成双层石墨烯结构.
Si3N4为六方相原子晶体,具有较高的热稳定性和介电性能.其介电常数为7,能带宽度为5.1 eV.具有较高的电阻率(1016Ω·cm)以及较高的介电强度(10 MV·cm−1),介电损耗小,并且与Si接触的界面良好.瑞典查尔姆斯理工大学的Sun等[79]尝试了1000°C下在Si3N4/Si上生长得到了2—70 nm厚的石墨烯薄膜.Liu实验组[80]优化了实验条件,利用两步生长的方法在Si3N4基底上生长石墨烯.首先向体系中引入较少的甲烷(2.3 sccm CH4,5 sccm H2,300 sccm Ar),诱发分立的石墨烯纳米晶的形成.将其作为成核点,继而引入较多的氢气(5 sccm CH4,50 sccm H2),实现石墨烯的平面生长,成功制备了具有高质量的单层的多晶石墨烯薄膜.其空穴的迁移率超过了1500 cm2·V−1·s−1,基本与金属催化生长的石墨烯质量相当.
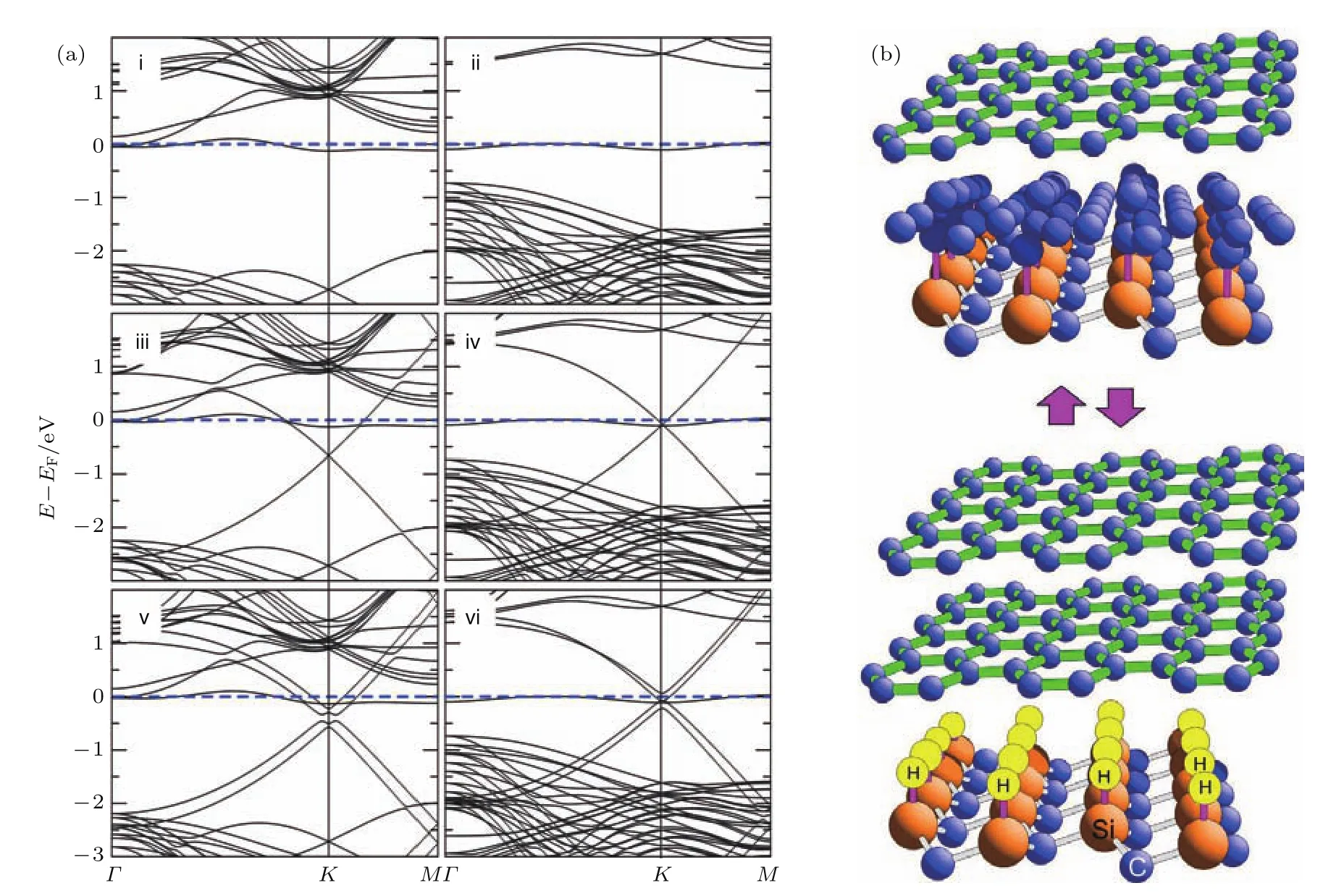
图10 (a)分别为在SiC表面生长(i),(ii)一层、(iii),(iv)两层以及(v),(vi)三层C薄膜的色散关系曲线[72];(b)利用可逆加氢反应将缓冲层C转化为石墨烯的原子结构示意图[73]Fig.10.(a)Dispersion curves for(i),(ii)mono-,(iii),(iv)bi-,(v),(vi)tri-layer C films on bulk SiC[72];(b)structure model of the reversible hydrogenation reaction,which transform the C bu ff er layer to sp2graphene layer[73].
3.4 玻 璃
近年来,石墨烯玻璃以其巨大的应用前景吸引了广泛的关注与研究.作为传统材料的代表,玻璃透光性好,价格低廉,性质稳定,在生产生活中具有十分广泛的应用.然而其无定型硅酸盐结构导致其导热及导电性能均较差,与石墨烯的结合可以弥补传统玻璃材料在电学应用领域的空白,如透明触摸屏、光电催化等.传统材料与新材料的结合,极大地扩宽了这两种材料的应用领域,加速了石墨烯规模化生产与实际应用的发展速率.
在这一领域,刘忠范院士所领导的团队进行了一系列的研究.玻璃不具备催化碳源的裂解的能力,并且在其表面C原子的迁移能力很弱.因而在玻璃上生长石墨烯需要在较高的温度下进行,以保证碳源的充分裂解,同时高温条件可以提高C原子的迁移能力以及吸附-成核的概率.他们首先探究了几种耐高温的玻璃材料(软化温度>1000°C):石英玻璃、硼硅玻璃和蓝宝石玻璃在1000°C时生长石墨烯的情况[81].研究发现,即使不采用任何金属催化剂辅助生长的条件下,通过优化生长参数,也可以在这些玻璃基底表面生长得到石墨烯薄膜.改变甲烷前驱物的浓度与生长时间(1—7 h),可以分别得到单层至多层的连续的石墨烯薄膜.如图11(a)中(i)—(iii)所示,该石墨烯薄膜具有良好的疏水性与透光性.拉曼谱图具有明显的石墨烯的信号峰;采用四探针法测试其薄层电阻为1.75—2.35 kΩ·sq−1(对于550 nm波长的入射光透光率为81%),载流子迁移率为553—710 cm2·V−1·s−1.

图11 (a)(i)在石英表面生长的石墨烯具有显著的疏水性;(ii)在覆盖了石墨烯的石英管中,液面为凸液面;(iii)展示了石墨烯玻璃的良好的透光性;(iv)为石墨烯在三种耐高温玻璃上的拉曼谱图;(v)为石墨烯薄膜的FET转移曲线;(vi)为石墨烯薄膜的透射光谱[81];(b)(i)在熔融态玻璃表面生长石墨烯的光学图片;(ii)在熔融态玻璃以及SiO2表面生长石墨烯的晶粒平面尺寸分布图,内插图分别展示了在两种基底上生长石墨烯的SEM图像[83];(c)利用APCVD与LPCVD在玻璃表面生长石墨烯的(i)示意图以及(ii)光学图片[85]Fig.11.(a)(i)Demonstration of the hydrophobic nature of graphene/quartz glass;(ii)photograph showing the convex surface of water contained in the graphene-coated tubes;(iii)photograph of graphene/sapphire glass with good transparency;(iv)Raman spectra of the grown graphene on three glasses;(v)transfer curve of the graphene FET;(vi)sheet resistance and UV-vis transmittance spectra of the graphene/quartz glass[81];(b)(i)photograph of the graphene grown on molten glass;(ii)SEM images and diameter distribution of the graphene disk on molten glass and SiO2/Si substrates[83];(c)(i)schematic diagram and(ii)photographs of the growth of graphene by APCVD and LPCVD process[85].
由于普通工业化玻璃如钠钙浮法玻璃高温稳定性较差,软化温度低至620°C,因而在1000°C左右的高温条件下无法将其作为生长石墨烯的基底.为克服石墨烯玻璃的基底的局限性,研究者利用前述的PECVD方法在相对低温的条件下(500—600°C)于商业化玻璃基底上实现了石墨烯薄膜的生长,进一步拓宽了石墨烯玻璃生长的窗口条件[82].与此同时,该组成员发现,在液态钠钙玻璃的表面也可以形成良好的石墨烯薄膜[83].将钠钙玻璃加热至1000°C时,其以熔融态的形式均匀铺展在石墨模具中.相较于固态基底,这种熔融态玻璃基底表面各向同性,减少了缺陷、扭折、粗糙等局部高能位点.并且在熔融态玻璃表面,C原子具有更高的迁移能力.因此在其表面石墨烯成核均匀,尺寸分布均一,且生长速率相对较快.如图11(b)所示,生长时间为1 h时,熔融玻璃表面形成了尺寸为0.8—1.1µm的圆盘形石墨烯.而在同样条件下,在固态二氧化硅表面生长得到的石墨烯晶粒随机分布,尺寸大小不一,晶粒尺寸仅在100—400 nm范围内.在最近的一篇报道中,熔融态也被用于大尺寸的二维过渡金属硫属化合物单晶结构[84].这充分展示了将二维材料的生长直接引入玻璃制造流程中,以实现其工业化生产的广大前景.
为了进一步提高石墨烯的生长质量,降低其薄层电阻同时提高电子输运性质,Liu课题组深入探究并优化了在石英上生长石墨烯的参数(较高的生长温度,较低的甲烷浓度以及较长的生长时间),并结合上述气态Cu原子催化生长石墨烯的方法,在石英表面制备了更高质量的石墨烯薄膜[29],其薄层电阻可低至370—510 Ω·sq−1(透光率为82%).通过瞬态吸收光谱可以直接推算出该石墨烯的载流子迁移率为4820 cm2·V−1·s−1,远远高于此前在玻璃以及其他介电层基底上生长石墨烯样品的迁移率.但这种方法也不可避免地引入了金属的残留,以及由金属蒸气梯度所引起的石墨烯生长不均匀的问题.
在这些报道中,石墨烯玻璃的尺寸均不超过4 in,并且石墨烯薄膜表面均匀度较差,生长周期较长(1—7 h).为了满足实际工业生产的需求,如何快速地实现超大面积的石墨烯玻璃的生长成为研究的重点.该课题组发现,改用乙醇作为石墨烯生长的碳源,在1100°C并且在低压环境中生长石墨烯时,可以在4 min内便在25 in的石英玻璃表面得到了均匀的石墨烯薄膜[85],如图11(c)所示.这一方面是由于相较于常压CVD体系,在低压环境中,管式炉内部的传质速率更快,并且在炉管内前驱物的浓度分布相对均匀,不存在较大的浓度梯度.另一方面是由于在温度高于800°C时,乙醇的分解十分完全,为石墨烯的生长供应了充足的C原子.因而在该工艺参数下,石墨烯薄膜的生长速率显著提高,并且该石墨烯玻璃表面十分均匀.这种工艺可以在短时间内制备得到大面积高质量、高均匀性的石墨烯玻璃,有望应用于实际工业生产中.
4 生长机理
在金属基底上生长石墨烯的生长机理目前研究得较为成熟,基本可以分为表面吸附生长与扩散析出生长两种生长机理.然而在介电层表面生长石墨烯的机理研究仍较为模糊.尽管已进行了大量的探究,但目前仍未提出一种普适的生长机理.这可能是由于在没有金属催化的情况下,石墨烯在介电衬底上的生长过程稳定性较差,对于生长环境、生长参数、生长基底等因素极为敏感,因而在不同的生长过程中,其生长机理并不完全一致.不考虑本文中所提到的金属辅助以及等离子体辅助法生长石墨烯的机理,目前已经报道的生长机理包括范德瓦耳斯外延生长、SiC纳米粒子催化生长、O原子催化生长等.
4.1 范德瓦耳斯外延生长
范德瓦耳斯外延生长是二维材料一种较为常见的生长模式.在这种生长模式下,外延的材料其晶格取向与基底的晶格取向保持固有的关系.这种方式所生长的石墨烯往往具有较高的均匀性与一致的晶体取向.该生长机理常见于与石墨烯具有一定程度的晶格匹配性的基底如h-BN,SiC,Al2O3等.图12(a)为典型的在h-BN表面外延生长石墨烯的AFM图像,在其表面可以观察到均匀的摩尔条纹[65].图12(b)形象地揭示了摩尔条纹的产生原因.研究发现,摩尔条纹的产生会对石墨烯产生一种周期性的超晶格势场,破坏其空间反演对称性,对其能带结构与性质带来新奇的现象,例如二级狄拉克点的衍生(如图12(c)所示)[63]、狄拉克点处带隙的打开[86]、零磁场中能谷拓扑电流的调控[87]、室温下的量子振荡效应[88]等.因而可控地通过范德瓦耳斯外延生长石墨烯对于基础量子理论研究、高性能电子器件以及相关领域的应用意义十分重大.另外,在SiC与Al2O3的表面石墨烯的生长也遵循范德瓦耳斯外延生长模式[51,71],如图11(d)与图11(e)所示.SiC表面外延石墨烯时表面晶格结构会发生一定改变,形成缓冲层.图11(d)中蓝色箭头所指为SiC基底所产生的衍射斑点,红色箭头指向表面生长石墨烯的衍射斑点,其余衍射斑点为缓冲层所产生.XPS谱图同样揭示了缓冲层C原子的存在.

图12 (a)h-BN表面外延生长石墨烯六边形单晶以及局部放大的图像[65];(b)在晶格取向一致时,由晶格失配产生的摩尔条纹的示意图;(c)h-BN表面外延生长取向一致的石墨烯的理论能带结构[63];(d)在SiC(0001)表面生长石墨烯的低能电子衍射(LEED)图像以及XPS光谱[71];(e)蓝宝石表面外延生长石墨烯的掠入式X射线衍射(GIXRD)谱图以及TEM图像[53]Fig.12.(a)A typical topography of a hexagonal graphene grains on h-BN and a zoom-in view[60];(b)illustration of the Moiré pattern generated by lattice mismatch with identical lattice orientation;(c)calculated band structure of epitaxial graphene on h-BN with zero rotation angle[58];(d)LEED pattern and XPS spectra of the graphene grown on SiC(0001)[84];(e)GIXRD image of the epitaxial graphene on sapphire substrate and TEM micrograph of a transferred graphene sheet[48].
4.2 SiC纳米粒子催化生长
在SiO2/Si基底上生长石墨烯的过程中,研究者发现,在高温下,C原子与表面的SiO2可能发生了热反应,生成了SiC粒子.在Qiu等[39]的报道中在石墨烯的生长过程中利用拉曼光谱检测到了SiC的存在,如图13(a),并提出SiC的存在可能催化了碳氢化合物裂解,促进了石墨烯的形成.Teng等[25]采用气态Cu原子催化生长石墨烯的实验中,利用XPS C 1s芯能级能谱图对石墨烯的生长过程进行分析演绎,结果如图13(b)所示.分析发现,在石墨烯生长的初期,具有三种化合态的C.其中,结合能为283.6 eV的主峰归属于SiC化合物,结合能为284.4 eV的峰为以sp2杂化方式键合的C原子,而285.3 eV处的峰归属于以sp3方式键合的C原子.随着生长时间的延长,SiC的峰强度逐渐减弱而sp2C原子的峰强度增大而成为主峰.说明在介电层上生成石墨烯的初期,在二氧化硅表面形成了部分SiC化合物,随着反应的进行,SiC比例逐渐减少,主要生成产物为石墨烯薄膜,充分证明SiC对于石墨烯的生长具有一定的催化作用.然而,在某些报道中,并未检测到SiC的存在;在SiC表面生长石墨烯也需要较高的温度(约1350°C)以维持其生长过程的进行,因而单纯的SiC可能并不具有显著的催化作用.可能的原因是,在SiO2生长石墨烯的初期所生成的SiC具有某种亚稳态相或特殊结构,因而催化了石墨烯的生长.目前SiC催化石墨烯生长的理论仍有待进一步的证实.

图13 在生长石墨烯过程中利用(a)拉曼谱图[39]以及(b)XPS谱图[25]观测到SiC纳米粒子的存在;(c)在蓝宝石表面生长石墨烯的机理示意图[51]Fig.13.(a)Raman spectra[39]and(b)XPS spectra[25]showing the existing of SiC during the growth of graphene;(c)schematic of the growth mechanism of graphene on sapphire[51].
高温下,C原子与Al2O3基底也会发生类似的热还原反应. Fanton等[50]开展的热力学计算与实验结果表明,在石墨烯的生长温度下(1425—1600°C),部分C原子会与Al2O3发生热还原反应,破坏Al2O3表面的光滑度,并且与石墨烯的生长构成竞争关系.图13(c)展示了在蓝宝石表面生长石墨烯的过程中,表面紧密堆积的O原子与C发生热还原反应释放出CO的生长机理[51].
4.3 O原子催化生长
O原子可以通过C—O键与H—O键的方式,提高对CHx小分子的俘获与结合能力,从而增加石墨烯成核概率与生长速率.因而一些含氧原子的介电材料可能具有较好的催化作用,如MgO,ZrO2[89],Al2O3[53],SrTiO3[55]等生长基底.对SiO2/Si基底进行空气退火也可以达到O催化生长石墨烯的效果.在其生长过程中,通过XPS测试可以检测到C—O键的存在,进一步可以证明O原子的耦合催化作用.
充分研究与理解在介电基底上生长石墨烯的生长机理对于制备高质量的石墨烯具有重要的指导意义.目前关于在介电层上生长石墨烯的机理的研究尚不充分,初步提出的一些理论模型亟待更加深入系统的理论与实验研究.
5 方法与质量评估
石墨烯的质量可以通过多种表征手段以及性能参数进行评估.在将石墨烯应用于微电子器件时,希望获得层数可控的、面积较大的、晶界或缺陷相对较少的石墨烯单晶或薄膜.表1中列举了2010年以来利用不同的生长手段以及采用不同的介电性/绝缘性的生长基底制备的石墨烯的晶粒尺寸、拉曼信号峰、生长速率、层数控制、透光率、薄层方阻、迁移率等性能参数.
由于实验条件的差异,采用不同的生长方法以及生长基底时,所制备的石墨烯质量各不相同.从表1中可以看到,在SiO2/Si基底表面生长的石墨烯迁移率平均为700 cm2·V−1·s−1左右,在SiC,Si3N4,Al2O3,SrTiO3表面生长的石墨烯迁移率基本可以达到103cm2·V−1·s−1,在h-BN表面生长的石墨烯甚至可以达到104cm2·V−1·s−1.不采用金属催化时,石墨烯的生长温度普遍较高(1000—1550°C),并且生长速率缓慢.金属辅助法对生长温度的要求一般在1000°C,这种方法制备的石墨烯具有金属残留,均匀性较差,这些因素可能导致其迁移率均低于103cm2·V−1·s−1,而PECVD法在较低的温度下即可以实现高质量石墨烯的生长.
6 总结与展望
在介电衬底上直接生长石墨烯具有重大的研究意义与应用前景,经过不断地研究与探索,已经取得了显著的成果.基于SiO2/Si,h-BN,SiC,Al2O3,SrTiO3以及玻璃等生长基底,发展出金属辅助法、PECVD法、热力学或动力学调控法等多种生长手段.通过优化生长参数,可制备出较大尺寸的石墨烯单晶以及晶圆级的石墨烯薄膜.石墨烯晶
体质量可以达到与在Cu箔基底上生长的石墨烯相当的程度.导电性与透光性完全满足透明导电电极的要求,成为替代ITO(氧化铟锡)的潜在材料.介电层上制备的石墨烯的载流子迁移率可以达到103—104cm2·V−1·s−1级别. 同时还可以直接实现直接掺杂以及图案化石墨烯的制备,为石墨烯集成电子器件的制备奠定了基础.

表1 2010 年以来在介电衬底表面生长石墨烯的生长基底、方法、以及各参数的总结Table 1. A summary of substrates, methods and performance of the synthesized graphene on dielectric substrates.
回顾近几年的研究进展,在介电层上生长石墨烯仍然存在一些问题以及难点.主要的挑战在于在介电层的表面生长石墨烯可控性相对较低.在石墨烯生长过程中,初始成核相对较难,成核点分布不均匀,成核密度以及生长速率等参数均难以控制.所生长的石墨烯具有明显的晶体缺陷,均匀性较差.大部分报道中单晶尺寸小于2µm,晶体质量以及尺寸仍有待提高.相信随着不断的探索与研究,在介电层上表面可生长得到高质量、大面积的石墨烯,并直接应用于微电子工业以及催化、能源等相关领域中.
[1]Novoselov K S,Geim A K,Morozov S V,Jiang D,Zhang Y,Dubonos S V,Grigorieva I V,Firsov A A 2004Science306 666
[2]Novoselov K S,Geim A K,Morozov S V,Jiang D,Katsnelson M I,Grigorieva I V,Dubonos S V,Firsov A A 2005Nature438 197
[3]Novoselov K S,Jiang Z,Zhang Y,Morozov S V,Stormer H L,Zeitler U,Maan J C,Boebinger G S,Kim P,Geim A K 2007Science315 1379
[4]Mayorov A S,Gorbachev R V,Morozov S V,Britnell L,Jalil R,Ponomarenko L A,Blake P,Novoselov K S,Watanabe K,Taniguchi T,Geim A K 2011Nano Lett.11 2396
[5]Katsnelson M I,Novoselov K S,Geim A K 2006Nat.Phys.2 620
[6]Guo W,Jing F,Xiao J,Zhou C,Lin Y,Wang S 2016Adv.Mater.28 3152
[7]Pan Y,Zhang H,Shi D,Sun J,Du S,Liu F,Gao H J 2009Adv.Mater.21 2777
[8]Lin Y C,Lu C C,Yeh C H,Jin C,Suenaga K,Chiu P W 2012Nano Lett.12 414
[9]Cheng Z,Zhou Q,Wang C,Li Q,Wang C,Fang Y 2011Nano Lett.11 767
[10]Suk J W,Lee W H,Lee J,Chou H,Piner R D,Hao Y,Akinwande D,Ruo ffR S 2013Nano Lett.13 1462
[11]Ambrosi A,Pumera M 2014Nanoscale6 472
[12]Suzuki S,Orofeo C M,Wang S,Maeda F,Takamura M,Hibino H 2013J.Phys.Chem.C117 22123
[13]Li X,Zhu Y,Cai W,Borysiak M,Han B,Chen D,Piner R D,Colombo L,Ruo ffR S 2009Nano Lett.9 4359
[14]Chen X,Wu B,Liu Y 2016Chem.Soc.Rev.45 2057
[15]Wang H,Yu G 2016Adv.Mater.28 4956
[16]Li X S,Cai W W,Colombo L,Ruo ffR S 2009Nano Lett.9 4268
[17]Levendorf M P,Ruiz-Vargas C S,Garg S,Park J 2009Nano Lett.9 4479
[18]Ismach A,Druzgalski C,Penwell S,Schwartzberg A,Zheng M,Javey A,Bokor J,Zhang Y 2010Nano Lett.10 1542
[19]Shin H J,Choi W M,Yoon S M,Han G H,Woo Y S,Kim E S,Chae S J,Li X S,Benayad A,Loc D D,Gunes F,Lee Y H,Choi J Y 2011Adv.Mater.23 4392
[20]Yan Z,Peng Z W,Sun Z Z,Yao J,Zhu Y,Zheng Liu,Ajayan P M,Tour J M 2011ACS Nano5 8187
[21]Tamaoki M,Imaeda H,Kishimoto S,Mizutani T 2013Appl.Phys.Lett.103 183114
[22]Xiong W,Zhou Y S,Jiang L J,Sarkar A,Mahjouri-Samani M,Xie Z Q,Gao Y,Ianno N J,Jiang L,Lu Y F 2013Adv.Mater.25 630
[23]Zhuo Q Q,Wang Q,Zhang Y P,Zhang D,Li Q L,Gao C H,Sun Y Q,Ding L,Sun Q J,Wang S D,Zhong J,Sun X H,Lee S T 2015ACS Nano9 594
[24]Kim K S,Zhao Y,Jang H,Lee S Y,Kim J M,Kim K S,Ahn J H,Kim P,Choi J Y,Hong B H 2009Nature457 706
[25]Teng P Y,Lu C C,Akiyama-Hasegawa K,Lin Y C,Yeh C H,Suenaga K,Chiu P W 2012Nano Lett.12 1379
[26]Kim H,Song I,Park C,Son M,Hong M,Kim Y,Kim J S,Shin H J,Baik J,Choi H C 2013ACS Nano7 6575
[27]Yen W C,Chen Y Z,Yeh C H,He J H,Chiu P W,Chueh Y L 2014Sci.Rep.4 4739
[28]Murakami K,Tanaka S,Hirukawa A,Hiyama T,Kuwajima T,Kano E,Takeguchi M,Fujita J I 2015Appl.Phys.Lett.106 093112
[29]Sun J,Chen Z,Yuan L,Chen Y,Ning J,Liu S,Ma D,Song X,Priydarshi M K,Bachmatiuk A,Rummeli M H,Ma T,Zhi L,Huang L,Zhang Y,Liu Z 2016ACS Nano10 11136
[30]Reina A,Thiele S,Jia X,Bhaviripudi S,Dresselhaus M S,Schaefer J A,Kong J 2009Nano Res.2 509
[31]Peng Z,Yan Z,Sun Z,Tour J M 2011ACS Nano5 8241
[32]Su C Y,Lu A Y,Wu C Y,Li Y T,Liu K K,Zhang W,Lin S Y,Juang Z Y,Zhong Y L,Chen F R,Li L J 2011Nano Lett.11 3612
[33]Kato T,Hatakeyama R 2012ACS Nano6 8508
[34]Kwak J,Chu J H,Choi J K,Park S D,Go H,Kim S Y,Park K,Kim S D,Kim Y W,Yoon E,Kodambaka S,Kwon S Y 2012Nat.Commun.3 645
[35]Wang J,Zeng M,Tan L,Dai B,Deng Y,Rummeli M,Xu H,Li Z,Wang S,Peng L,Eckert J,Fu L 2013Sci.Rep.3 2670
[36]Tan L F,Zeng M Q,Zhang T,Fu L 2015Nanoscale7 9105
[37]Berman D,Deshmukh S A,Narayanan B,Sankaranarayanan S K,Yan Z,Balandin A A,Zinovev A,Rosenmann D,Sumant A V 2016Nat.Commun.7 12099
[38]Zhang L,Shi Z,Wang Y,Yang R,Shi D,Zhang G 2011Nano Res.4 315
[39]Medina H,Lin Y C,Jin C,Lu C C,Yeh C H,Huang K P,Suenaga K,Robertson J,Chiu P W 2012Adv.Funct.Mater.22 2123
[40]Wei D,Lu Y,Han C,Niu T,Chen W,Wee A T 2013Angew.Chem.Int.Ed.Engl.52 14121
[41]Wei D C,Peng L,Li M L,Mao H Y,Niu T C,Han C,Chen W,Wee A T S 2015ACS Nano9 164
[42]Kim Y S,Joo K,Jerng S K,Lee a H,Moon D,Kim j,Yoon E,Chun S H 2014ACS Nano8 2230
[43]Kim Y S,Joo K,Jerng S K,Lee J H,Yoon E,Chun S H 2014Nanoscale6 10100
[44]Hao Y,Bharathi M S,Wang L,Liu Y,Chen H,Nie S,Wang X,Chou H,Tan C,Fallahazad B,Ramanarayan H,Magnuson C W,Tutuc E,Yakobson B I,McCarty K F,Zhang Y W,Kim P,Hone J,Colombo L,Ruo ffR S 2013Science342 720
[45]Chen J,Wen Y,Guo Y,Wu B,Huang L,Xue Y,Geng D,Wang D,Yu G,Liu Y 2011J.Am.Chem.Soc.133 17548
[46]Behura S,Nguyen P,Che S,Debbarma R,Berry V 2015J.Am.Chem.Soc.137 13060
[47]Chen J,Guo Y,Jiang L,Xu Z,Huang L,Xue Y,Geng D,Wu B,Hu W,Yu G,Liu Y 2014Adv.Mater.26 1348
[48]Pang J,Mendes R G,Wrobel P S,Wlodarski M D,Ta H Q,Zhao L,Giebeler L,Trzebicka B,Gemming T,Fu L,Liu Z,Eckert J,Bachmatiuk A,Rummeli M H 2017ACS Nano11 1946
[49]Hwang J,Shields V B,Thomas C I,Shivaraman S,Hao D,Kim M,Woll A R,Tompa G S,Spencer M G 2010J.Cryst.Growth312 3219
[50]Fanton M A,Robinson J A,Puls C,Liu Y,Hollander M J,Weiland B E,LaBella M,Trumbull K,Kasarda R,Howsare C,Stitt J,Snyder D W 2011ACS Nano5 8062
[51]Hwang J,Kim M,Campbell D,Alsalman H A,Kwak J Y,Shivaraman S,Woll A R,Singh A K,Hennig R G,Gorantla S,mmeli M H R,Spencer M G 2013ACS Nano7 385
[52]Miyasaka Y,Nakamura A,Temmyo J 2011Jpn.J.Appl.Phys.50 04DH12
[53]Song H J,Son M,Park C,Lim H,Levendorf M P,Tsen A W,Park J,Choi H C 2012Nanoscale4 3050
[54]Park J,Lee J,Choi J H,Hwang D K,Song Y W 2015Sci.Rep.5 11839
[55]Sun J,Gao T,Song X,Zhao Y,Lin Y,Wang H,Ma D,Chen Y,Xiang W,Wang J,Zhang Y,Liu Z 2014J.Am.Chem.Soc.136 6574
[56]Li X A,Liu Z R,Wang B L,Yang J P,Ma Y W,Feng X M,Huang W,Gu M F 2013Synth.Met.174 50
[57]Rümmeli M H,Bachmatiuk A,Scott A,Börrnert F,Warner J H,Ho ff man V,Lin J H,Cuniberti G,Büchner B 2010ACS Nano4 4206
[58]Young A F,Dean C R,Meric I,Sorgenfrei S,Ren H,Watanabe K,Taniguchi T,Hone J,Shepard K L,Kim P 2012Phys.Rev.B85 235458
[59]Ding X,Ding G,Xie X,Huang F,Jiang M 2011Carbon49 2522
[60]Tang S,Ding G,Xie X,Chen J,Wang C,Ding X,Huang F,Lu W,Jiang M 2012Carbon50 329
[61]Son M,Lim H,Hong M,Choi H C 2011Nanoscale3 3089
[62]Mishra N,Miseikis V,Convertino D,Gemmi M,Piazza V,Coletti C 2016Carbon96 497
[63]Yang W,Chen G,Shi Z,Liu C C,Zhang L,Xie G,Cheng M,Wang D,Yang R,Shi D,Watanabe K,Taniguchi T,Yao Y,Zhang Y,Zhang G 2013Nat.Mater.12 792
[64]Yankowitz M,Xue J,Cormode D,Sanchez-Yamagishi J D,Watanabe K,Taniguchi T,Jarillo-Herrero P,Jacquod P,LeRoy B J 2012Nat.Phys.8 382
[65]Tang S,Wang H,Zhang Y,Li A,Xie H,Liu X,Liu L,Li T,Huang F,Xie X,Jiang M 2013Sci.Rep.3 2666
[66]Wang M,Jang S K,Jang W J,Kim M,Park S Y,Kim S W,Kahng S J,Choi J Y,Ruo ffR S,Song Y J,Lee S 2013Adv.Mater.25 2746
[67]Gao T,Song X,Du H,Nie Y,Chen Y,Ji Q,Sun J,Yang Y,Zhang Y,Liu Z 2015Nat.Commun.6 6835
[68]Tang S,Wang H,Wang H S,Sun Q,Zhang X,Cong C,Xie H,Liu X,Zhou X,Huang F,Chen X,Yu T,Ding F,Xie X,Jiang M 2015Nat.Commun.6 6499
[69]Li Q C,Zhao Z F,Yan B M,Song X J,Zhang Z P,Li J,Wu X S,Bian Z Q,Zou X L,Zhang Y F,Liu Z F 2017Adv.Mater.29 1701325
[70]Berger C,Song M Z,Li X B,Wu X S,Brown N,Naud C C,Mayou D,Li T B,Hass J,Marchenkov A N,Conrad E H,First P N,Heer W A D 2006Science312 1191
[71]Emtsev K V,Bostwick A,Horn K,Jobst J,Kellogg G L,Ley L,McChesney J L,Ohta T,Reshanov S A,Rohrl J,Rotenberg E,Schmid A K,Waldmann D,Weber H B,Seyller T 2009Nat.Mater.8 203
[72]Varchon F,Feng R,Hass J,Li X,Nguyen B N,Naud C,Mallet P,Veuillen J Y,Berger C,Conrad E H,Magaud L 2007Phys.Rev.Lett.99 126805
[73]Virojanadara C,Zakharov A A,Yakimova R,Johansson L I 2010Surf.Sci.604 L4
[74]Walter A L,Jeon K J,Bostwick A,Speck F,Ostler M,Seyller T,Moreschini L,Kim Y S,Chang Y J,Horn K,Rotenberg E 2011Appl.Phys.Lett.98 184102
[75]Ostler M,Fromm F,Koch R J,Wehrfritz P,Speck F,Vita H,Böttcher S,Horn K,Seyller T 2014Carbon70 258
[76]Virojanadara C,Watcharinyanon S,Zakharov A A,Johansson L I 2010Phys.Rev.B82 205402
[77]Xia C,Watcharinyanon S,Zakharov A A,Yakimova R,Hultman L,Johansson L I,Virojanadara C 2012Phys.Rev.B85 045418
[78]Emtsev K V,Zakharov A A,Coletti C,Forti S,Starke U 2011Phys.Rev.B84 125423
[79]Sun J,Lindvall N,Cole M T,Yurgens A 2011Appl.Phys.Lett.98 252107
[80]Chen J,Guo Y,Wen Y,Huang L,Xue Y,Geng D,Wu B,Luo B,Yu G,Liu Y 2013Adv.Mater.25 992
[81]Sun J,Chen Y,Priydarshi M K,Chen Z,Bachmatiuk A,Zou Z,Chen Z,Song X,Gao Y,Rummeli M H,Zhang Y,Liu Z 2015Nano Lett.15 5846
[82]Sun J,Chen Y,Cai X,Ma B,Chen Z,Priydarshi M K,Chen K,Gao T,Song X,Ji Q,Guo X,Zou D,Zhang Y,Liu Z 2015Nano Res.8 3496
[83]Chen Y,Sun J,Gao J,Du F,Han Q,Nie Y,Chen Z,Bachmatiuk A,Priydarshi M K,Ma D,Song X,Wu X,Xiong C,Rummeli M H,Ding F,Zhang Y,Liu Z 2015Adv.Mater.27 7839
[84]Chen J,Zhao X,Tan S J,Xu H,Wu B,Liu B,Fu D,Fu W,Geng D,Liu Y,Liu W,Tang W,Li L,Zhou W,Sum T C,Loh K P 2017J.Am.Chem.Soc.139 1073
[85]Chen X D,Chen Z,Jiang W S,Zhang C,Sun J,Wang H,Xin W,Lin L,Priydarshi M K,Yang H,Liu Z B,Tian J G,Zhang Y,Zhang Y,Liu Z 2017Adv.Mater.29 1603428
[86]Wang E,Lu X,Ding S,Yao W,Yan M,Wan G,Deng K,Wang S,Chen G,Ma L,Jung J,Fedorov A V,Zhang Y,Zhang G,Zhou S 2016Nat.Phys.12 1111
[87]Gorbachev R V,Song J C W,Yu G L,Kretinin A V,Withers F,Cao Y,Mishchenko A,Grigorieva I V,Novoselov K S,Levitov L S,Geim A K 2014Science346 448
[88]Krishna Kumar R,Chen X,Auton G H,Mishchenko A,Bandurin D A,Morozov S V,Cao Y,Khestanova E,Ben Shalom M,Kretinin A V,Novoselov K S,Eaves L,Grigorieva I V,Ponomarenko L A,Fal’ko V I,Geim A K 2017Science357 181
[89]Scott A,Dianat A,Börrnert F,Bachmatiuk A,Zhang S,Warner J H,Borowiak-Paleń E,Knupfer M,Büchner B,Cuniberti G,Rümmeli M H 2011Appl.Phys.Lett.98 073110
PACS:68.65.Pq,81.15.Gh,77.55.—g DOI:10.7498/aps.66.216804
*Project supported by the National Basic Research Program of China(Grant No.2013CB632900)and the National Natural Science Foundation of China(Grant Nos.61390502,21373068).
†Corresponding author.E-mail:hupa@hit.edu.cn
Research progress of direct synthesis of graphene on dielectric layer∗
Yang Hui-Hui1)2)Gao Feng1)2)Dai Ming-Jin1)2)Hu Ping-An1)2)†
1)(School of Materials and Science Engineering,Harbin Institute of Technology,Harbin 150080,China)
2)(Key Lab of Microsystem and Microstructure(Ministry of Education),Harbin Institute of Technology,Harbin 150080,China)
d 5 August 2017;revised manuscript
26 September 2017)
As one of the most appealing materials,graphene possesses remarkable electric,thermal,photoelectric and mechanic characteristics,which make it extremely valuable both for fundamental researches and practical applications.Nowadays the synthesis of graphene is commonly achieved by growing on metal substrate via chemical vapor deposition.For the integration in micro-electric device,the as-grown graphene needs to be transferred onto target dielectric layer.However,wrinkles,cracks,damages,and chemical residues from the metal substrate and the auxiliary polymer are inevitably introduced to graphene during such a transfer process,which are greatly detrimental to the performances of the graphene devices.Therefore,the direct synthesis of graphene on dielectric layer is of great importance.Many researches about this subject have been carried out in the last few years.While only few papers have systematically reviewed the direct growth of graphene on dielectric layer.For the in-depth understanding and further research of it,a detailed overview is required.In this paper,we summarize the recent research progress of the direct syntheses of graphene on dielectric layers,and expatiate upon different growth methods,including metal assisted growth,plasma enhanced growth,thermodynamics versus kinetics tailored growth,et al.Then di ff erences in property between graphenes grown on various dielectric and insulating layers which serve as growth substrates in the direct growing process are discussed,such as SiO2/Si,Al2O3,SrTiO3,h-BN,SiC,Si3N4and glass.Some kinds of mechanisms for graphene to be directly grown on dielectric layers have been proposed in different reports.Here in this paper,we review the possible growth mechanisms and divide them into van der Waals epitaxial growth and catalytic growth by SiC nanoparticles or oxygen atoms.Detailed data including Raman signals,sheet resistances,transmittances,carrier motilities are listed for the direct comparison of the quality among the graphenes grown on dielectric layers.The research focus and major problems existing in this field are presented in the last part of this paper.We also prospect the possible developing trend in the direct syntheses of high quality graphenes on dielectric layers in the future.
graphene,dielectric layer,chemical vapor deposition,transfer-free
作为21世纪备受瞩目的材料,石墨烯兼具优异的电、热、光与力学性质,具有十分广阔的研究价值与应用价值.目前主要通过在金属基底上生长获得石墨烯,并将其转移至目标介电层基底上以构筑电子器件.转移过程不可避免地引入了褶皱、裂纹、破损以及聚合物/金属残留,严重损害了石墨烯的性能.因而直接在介电基底上制备高质量的石墨烯薄膜具有重要意义.本文总结了近年来在介电衬底上直接生长石墨烯的研究进展:阐述了金属辅助法、等离子体增强法以及热力学或动力学调控法等多种生长手段;介绍了多种介电/绝缘基底包括SiO2/Si,Al2O3,SrTiO3,h-BN,SiC,Si3N4以及玻璃表面生长石墨烯的特点与性能,分析了其可能的生长机理.根据拉曼谱图、薄层电阻、透光率、载流子迁移率等评估指标,将多种方法得到的石墨烯质量进行了总结与比较,并提出了直接在介电衬底上生成石墨烯的研究难点与趋势.
10.7498/aps.66.216804
∗国家重点基础研究发展计划(批准号:2013CB632900)和国家自然科学基金(批准号:61390502,21373068)资助的课题.
†通信作者.E-mail:hupa@hit.edu.cn
©2017中国物理学会Chinese Physical Society
